↧
Drug Patents International
↧
Janssen seeks FDA approval for Yondelis (Trabectedin) drug to treat advanced STS
Janssen seeks FDA approval for Yondelis (Trabectedin) drug to treat advanced STS

ET-743, Yondelis (trabectedin)
Trabectedin, Ecteinascidin 743, NSC-684766, ET-743, Yondelis, ID0YZQ2TCP
cas 114899-77-3
(-)-(1’R,6R,6aR,7R,13S,14S,16R)-5-Acetoxy-6′,8,14-trihydroxy-7′,9-dimethoxy-4,10,23-trimethyl-1′,2′,3′,4′,6a,7,12,13,14,16-decahydro-6H-spiro[6,16-(epithiopropanoxymethano)-7,13-epimino-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′-isoquinolin]-19-one
Janssen seeks FDA approval for Yondelis drug to treat advanced STS
Janssen Research & Development is seeking approval from US Food and Drug Administration (FDA) for its Yondelis (trabectedin) to treat patients with advanced soft tissue sarcoma (STS).
Trabectedin, also referred as ET-743 during its development, is a marine derived antitumoral agent discovered in the Carribean tunicate _Ecteinascidia turbinata_ and now produced synthetically. Trabectedin has a unique mechanism of action. It binds to the minor groove of DNA interfering with cell division and genetic transcription processes and DNA repair machinery.It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have granted orphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Trabectedin (also known as ecteinascidin 743 or ET-743) is an anti-tumor drug. It is sold byZeltia and Johnson and Johnson under the brand name Yondelis. It is approved for use in Europe, Russia and South Korea for the treatment of advanced soft tissue sarcoma. It is also undergoing clinical trials for the treatment of breast, prostate, and paediatric sarcomas. The European Commission and the U.S. Food and Drug Administration (FDA) have grantedorphan drug status to trabectedin for soft tissue sarcomas and ovarian cancer.
Discovery and development
The ecteinascidins (herein abbreviated ETs) are exceedingly potent antitumor agents isolated from the marine tunicate Ecteinascidia turbinata. Several ecteinascidins have been reported previously in the patent and scientific literature. See, for example U.S. Pat. No. 5,089,273, which describes novel compounds of matter extracted from the tropical marine invertebrate Ecteinascidia turbinata, and designated therein as ecteinascidins 729, 743, 745, 759A, 759B and 770. These compounds are useful as antibacterial and/or antitumor agents in mammals. U.S. Pat. No. 5,478,932 describes other novel ecteinascidins isolated from the Caribbean tunicate Ecteinascidia turbinata, which provide in vivo antitumor activity against P388 lymphoma, B16 melanoma, M5076 ovarian sarcoma, Lewis lung carcinoma, and the LX- I human lung and MX- 1 human mammary carcinoma xenografts.
One of the ETs, ecteinascidin 743 (ET-743), is a tetrahydroisoquinoline alkaloid with considerable in vitro and in vivo antitumor activity in murine and human tumors, and potent antineoplastic activity against a variety of human tumor xenografts grown in athymic mice, including melanoma, ovarian and breast carcinoma.
ET-743 is a natural compound with the following structure:

ET-743 is also known with the generic name trabectedin and the trademark Yondelis®, and it is currently approved in Europe for the treatment of soft tissue sarcoma. The clinical development of trabectedin continues in phase 11/ III clinical trials in breast, ovarian and prostate cancer. A clinical development program of ET-743 in cancer patients was started with phase I studies investigating 1- hour, 3-hour, 24-hour, and 72-hour intravenous infusion schedules and a 1 hour daily x 5 (dx5) schedule. Promising responses were observed in patients with sarcoma, breast and ovarian carcinoma.
Therefore this new drug is currently under intense investigation in several phase 11/ III clinical trials in cancer patients with a variety of neoplastic diseases. Further information regarding the dosage, schedules, and administration of ET-743 for the treatment of cancer in the human body, either given alone or in combination is provided in WO 00/69441 , WO 02/36135, WO 03/39571 , WO 2004/ 105761 , WO 2005/039584, WO 2005/049031 , WO 2005/049030, WO 2005/049029, WO 2006/046080, WO 2006/005602, and PCT/US07/98727, which are incorporated by reference herein in their entirety.
A review of ET-743, its chemistry, mechanism of action and preclinical and clinical development can be found in Kesteren, Ch.
Van et al., Anti-Cancer Drugs, 2003, 14 (7), 487-502: “ET-743 (trabectedin, ET-743): the development of an anticancer agent of marine origin”, and references therein.
During the past 30 years medical oncologists have focused to optimise the outcome of cancer patients and it is just now that the new technologies available are allowing to investigate polymorphisms, gene expression levels and gene mutations aimed to predict the impact of a given therapy in different groups of cancer patients to tailor chemotherapy. Representative examples include the relationship between the Thymidylate Synthase (TS) mRNA expression and the response and the survival with antifolates, beta tubulin III mRNA levels and response to tubulin interacting agents, PTEN gene methylation and resistance to CPT- I l and, STAT3 over expression and resistance to Epidermal Growth Factor (EGF) interacting agents.
A molecular observation of potential clinical impact relates to the paradoxical relation between the efficiency of the Nucleotide Excision Repair (NER) pathway and the cytotoxicity of ET-743. In fact, tumour cells that are efficient in this DNA repair pathway appear to be more sensitive to ET-743. This evidence is in contrast with the pattern noted with platin based therapeutic regimens which are highly dependent on the lack of activity of this repair pathway (ie. an increase in ERCCl expression has been associated to clinical resistance to platinum-based anti-cancer therapy).
There are evidences on the key role of NER pathways on the cytotoxicity of ET-743 in cell lines. ET-743 binds to G residues in the minor groove of DNA forming adducts that distort the DNA helix structure and they are recognised by NER mechanisms (Pourquier, P. et al., 2001 , Proceedings of the American Association for Cancer Research Annual Meeting, Vol. 42, pp. 556. 92nd Annual Meeting of the American Association for Cancer Research. New Orleans, LA, USA. March 24-28, 2001. ISSN: 0197-016X). Takebayasi et al. (Nature Medicine, 2001 , 7(8), 961-966) have proposed that the presence of these DNA adducts in transcribed genes, blocks the Transcription Coupled NER (TC-NER) system by stalling the cleavage intermediates and producing lethal Single Strand Breaks (SSBs). It is known from Grazziotin et al (Proc.Natl.Acad.Sic.USA, 104: 13062- 13067) that the DNA adducts formed by exposure to ET-743 are transformed into double strand DNA breaks.
The fact that NER mediates ET-743 ‘s cytotoxicity has also been found in the yeast Saccharomyces cerevisae by Grazziotin et al. (Biochemical Pharmacology, 2005, 70, 59-69) and in the yeast Schizosaccharomyces pombe by Herrero et al. (Cancer Res. 2006, 66(16), 8155-8162).
In addition, Bueren et al. (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have been shown that ET-743 induces double-strand breaks in the DNA in early S phase that are detected and repaired by the Homologous Recombination Repair (HRR) pathway. In addition, Erba et al (Eur. J. Cancer, 2001 , 37(1), 97- 105) and Bueren et al (Proceedings AACR Annual Meeting 2007, Abstract no. 1965) have shown that inactivation/ mutations of genes related to the Double Strand Break detection such as DNA-PK, ATM and ATR and of genes related to Homologous Recombination Repair pathway, such as Fanconi Anemia genes, BRCAl , BRCA2 and RAD51 make cells more sensitive to trabectedin. Such unique finding is the opposite to the pattern with conventional DNA interacting agents, like in the case of microtubule poisons such as taxanes and vinorelbine.
Finally, pharmacogenomic studies prior have demonstrated that increased expression of the NER genes ERCCl and XPD in the tumor tissue does not impact the outcome of patients treated with
ET-743. However, the low expression of BRCAl in the tumor tissue is correlated with a better outcome in cancer patents treated with
ET-743. Further information can be found in WO 2006/005602, which is incorporated by reference herein in its entirety.
Three rare, autosomal recessive inherited human disorders are associated with impaired NER activity: xeroderma pigmentosum (XP), Cockayne Syndrome (CS), and trichothiodystrophy (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245- 274). XP patients exhibit extreme sensitivity to sunlight, resulting in a high incidence of skin cancers (Kraemer et al. Arch. Dermatol. 123, 241-250, and Arch. Dermatol. 130, 1018- 1021). About 20% of XP patients also develop neurologic abnormalities in addition to their skin problems. These clinical findings are associated with cellular defects, including hypersensitivity to killing and mutagenic effects of UV, and inability of XP cells to repair UV-induced DNA damage (van Steeg et al. MoI. Med. Today, 1999, 5, 86-94).
Seven different NER genes, which correct seven distinct genetic XP complementation groups (XPA-XPG), have been identified (Bootsma et al. The Genetic Basis of Human Cancer. McGraw-Hill, 1998, 245-274). The human gene responsible for XP group G was identified as ERCC5 (Mudgett et al. Genomics, 1990, 8, 623-633; O’Donovan et al. Nature, 1993, 363, 185- 188; and Nouspikel et al. Hum. MoI. Genet. 1994, 3, 963-967). The XPG gene codes for a structure-specific endonuclease that cleaves damaged DNA ~5 nt 3′ to the site of the lesion and is also required non-enzymatically for subsequent 5′ incision by the XPF/ ERCCl heterodimer during the NER process (Aboussekhra et al. Cell, 1995, 80, 859-868; Mu et al. J. Biol. Chem. 1996, 271 , 8285-8294; and Wakasugi et al. J. Biol. Chem. 1997, 272, 16030- 16034). There is also evidence suggesting that XPG is also involved in transcription-coupled repair of oxidative DNA lesions (Le Page et al. Cell, 101 , 159- 171).
Takebayashi et al. (Cancer Lett., 2001 , 174: 1 15- 125) have observed an increase in heterozygosity loss and microsatellite instability in a substantial percentage of samples of ovarian, lung and colon carcinoma. Le Moirvan et al, (Int.J. Cancer, 2006,1 19: 1732- 1735) have described the presence of polymorphisms in the XPG gene in sarcoma patients. It is also known from Takebayashi et al. (Proceedings of the American Association forCancer Research Annual Meeting, March, 2001 , Vol. 42, pp. 813.92nd Annual Meeting of the American Association for Cancer
Research. New Orleans, LA, USA. March 24-28, 2001) that cells deficient in the NER system are resistant to treatment with ET-743 (Zewail-Foote, M. et al., 2001 , Chemistry and Biology, 8: 1033- 1049 and Damia, G. et al., 2001 , Symposium AACR NCI EORTC) and that the antiproliferative effects of ET-743 require a functional XPG gene.
Since cancer is a leading cause of death in animals and humans, several efforts have been and are still being undertaken in order to obtain an antitumor therapy active and safe to be administered to patients suffering from a cancer. Accordingly, there is a need for providing additional antitumor therapies that are useful in the treatment of cancer.
Trabectedin is a tetrahydroisoquinoline, a novel marine-derived antitumor agent isolated from the colonial tunicate Ecteinascidia turbinate. The drug binds to the minor groove of the DNA, bending the DNA towards the major groove, blocking the activation of genes in a unique way via several pathways, including selective inhibition of the expression of key genes (including oncogenes) involved in cell growth and drug resistance, inhibition of genetic repair pathways and inhibition of cell cycle progression leading to p53-independent programmed cell death.
In July 2003, the European Committee of Proprietary Medicinal Products (CPMP) recommended against granting marketing authorization to trabectedin for soft tissue sarcoma. PharmaMar appealed the decision in September 2003. Later that year, the CPMP rejected the company’s appeal. In 2006, the company filed another regulatory application for this indication and, finally, in 2007, a positive opinion was received in the E.U. for the treatment of metastatic soft tissue sarcoma. First commercialization of the product in the E.U. took place in October 2007 in the U.K. and Germany.
The compound is also available in several other countries. In 2008, the compound was filed for approval in the U.S. and the E.U. for the treatment of relapsed advanced ovarian cancer in combination with liposomal doxorubicin, and in 2009 approval was received in both countries. Trabectedin is available in several European countries, including the U.K. and Germany. Also in 2009 the drug candidate was approved in Philippines for the ovarian cancer indication.
The compound had been in phase II development by Johnson & Johnson for the treatment of prostate cancer; however, no recent development has been reported for this research. PharmaMar is evaluating the compound in phase II trials for the treatment of breast cancer. Additional early clinical trials are ongoing at the National Cancer Institute (NCI) to evaluate trabectedin for potential use in the treatment of advanced, persistent or recurrent uterine leiomyosarcomas and solid tumors.
In 2011, a regulatory application that had been filed in the U.S. seeking approval for the treatment of relapsed advanced disease in combination with liposomal doxorubicin was withdrawn by the company based on the FDA’s recommendation that an additional phase III study be conducted to obtain approval. In 2014, Janssen Research & Development, LLC submitted an NDA for trabectedin to the FDA for the treatment of patients with advanced soft tissue sarcoma (STS), including liposarcoma and leiomyosarcoma subtypes, who have received prior chemotherapy including an anthracycline.
Trabectedin was developed by PharmaMar, a subsidiary of Zeltia. The drug was being codeveloped and comarketed in partnership with Ortho Biotech, a subsidiary of Johnson & Johnson pursuant to an agreement signed in 2001. However, in 2008 the license agreement between the two companies was terminated.
The compound was granted orphan drug designation for the treatment of soft tissue sarcoma and for the treatment of ovarian cancer by the FDA and the EMEA. In 2011, orphan drug designation was granted in Japan for the treatment of malignant soft tissue tumor accompanied with chromosomal translocation. In 2009, the product was licensed to Taiho by PharmaMar in Japan for the treatment of cancer.
During the 1950s and 1960s, the National Cancer Institute carried out a wide ranging program of screening plant and marine organism material. As part of that program extract from the sea squirt Ecteinascidia turbinata was found to have anticancer activity in 1969.[1]Separation and characterisation of the active molecules had to wait many years for the development of sufficiently sensitive techniques, and the structure of one of them, Ecteinascidin 743, was determined by KL Rinehart at the University of Illinois in 1984.[2]Rinehart had collected his sea squirts by scuba diving in the reefs of the West Indies.[3]
Recently, the biosynthetic pathway responsible for producing the drug, has been determined to come from Candidatus Endoecteinascidia frumentensis, a microbial symbiont of the tunicate.[4] The Spanish company PharmaMar licensed the compound from the University of Illinois before 1994 and attempted to farm the sea squirt with limited success.[3]
Yields from the sea squirt are extremely low – it takes 1 tonne of animals to isolate 1 gram of trabectedin – and about 5 grams were believed to be needed for a clinical trial[5] so Rinehart asked the Harvard chemist E. J. Corey to search for a synthetic method of preparation. His group developed such a method and published it in 1996.[6] This was later followed by a simpler and more tractable method which was patented by Harvard and subsequently licensed to PharmaMar.[3] The current supply is based on a semisynthetic process developed by PharmaMar starting from Safracin B, an antibiotic obtained by fermentation of the bacterium Pseudomonas fluorescens.[7] PharmaMar have entered into an agreement with Johnson and Johnson to market the compound outside Europe.
Trabectedin was first dosed in humans in 1996.In 2007, the EMEA gave authorisation for the marketing of trabectedin, under the trade name Yondelis, for the treatment of patients with advanced soft tissue sarcoma, after failure of anthracyclines and ifosfamide, or who are unsuited to receive these agents. The agency’s evaluating committee, the CHMP observed that trabectedin had not been evaluated in an adequately designed and analyzed randomized trial against current best care, and that the clinical efficacy data was mainly based on patients with liposarcoma and leiomyosarcoma. However the pivotal study did show a significant difference between two different trabectedin treatment regimens, and due to the rarity of the disease the CHMP considered that marketing authorisation could be granted under exceptional circumstances.[8] As part of the approval PharmaMar agreed to conduct a further trial to identify whether any specific chromosomal translocations could be used to predict responsiveness to trabectedin.[9] Trabectedin is also approved in South Korea[10] and Russia.
In 2008 the submission was announced of a registration dossier to the European Medicines Agency (EMEA) and the FDA for Yondelis when administered in combination with pegylated liposomal doxorubicin (Doxil, Caelyx) for the treatment of women with relapsed ovarian cancer. In 2011, Johnson&Johnson voluntarily withdrew the submission in the United States following a request by the FDA for an additional Phase III study to be done in support of the submission.[11]
Trabectedin is also in phase II trials for prostate, breast and paediatric cancers.[12]
Structure

Trabectedin is composed of 3 tetrahydroisoquinoline moieties, 8 rings including one 10-membered heteocyclic ring containing a cysteine residue, and 7 chiral centers.
Biosynthesis
The biosynthesis of Trabectedin in Candidatus Endoecteinascidia frumentensis starts with a fatty acid loading onto the acyl-ligase domain of the EtuA3 module. A cysteine and glycine are then loaded as canonical NRPS amino acids. A tyrosine residue is modified by the enzymes EtuH, EtuM1, and EtuM2 to add a hydroxyl at the meta position of the phenol, and adding two methyl groups at the para-hydroxyl and the meta carbon position. This modified tyrosine reacts with the original substrate via a Pictet-Spangler reaction, where the amine group is converted to an imine by deprotonation, then attacks the free aldehyde to form a carbocation that is quenched by electrons from the methyl-phenol ring. This is done in the EtuA2 T-domain. This reaction is done a second time to yeid a dimer of modified tyrosine residues that have been further cyclized via Pictet-spangler reaction, yielding a bicyclic ring moiety. The EtuO and EtuF3 enzymes continue to post-translationally modify the molecule, adding several functional groups and making a sulfide bridge between the original cysteine residue and the beta-carbon of the first tyrosine to form ET-583, ET-597, ET-596, and ET-594 which have been previously isolated.[4] A third o-methylated tyrosine is added and cyclized via Pictet-Spangler to yield the final product.[4]

Proposed biosynthetic scheme for the biosynthesis of Trabecteden (ET-743)
Synthesis
The total synthesis by E.J. Corey used this proposed biosynthesis to guide their synthetic strategy. The synthesis uses such reactions as the Mannich reaction, Pictet-Spengler reaction, the Curtius rearrangement, and chiral rhodium-based diphosphine-catalyzedenantioselective hydrogenation. A separate synthetic process also involved the Ugi reaction to assist in the formation of the pentacyclic core. This reaction was unprecedented for using such a one pot multi-component reaction in the synthesis of such a complex molecule.
Org Lett 2000,2(7),993
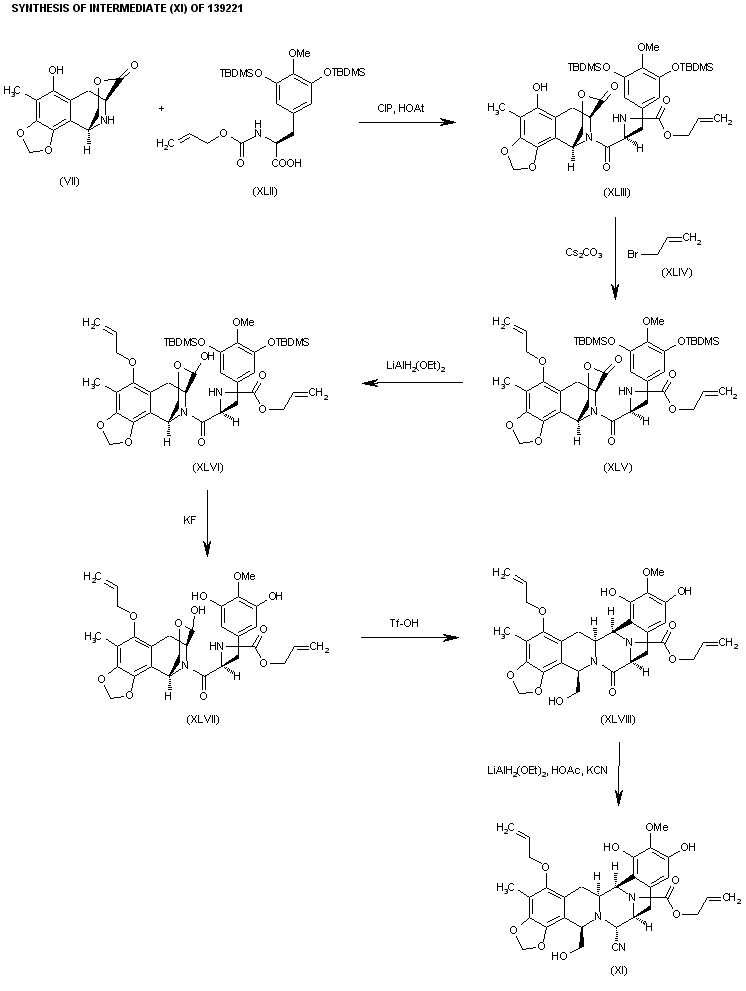
The previously reported synthesis of 139221 (scheme 13922101a) has been investigated in order to find a more efficient, reproducible and economical route to work in the mutikilogram scale. Herein it is reported a new process which is simpler and proceeds with an overall yield of 54% (the original process, 35%). The condensation of intermediate aminolactone (I) (scheme 13922101a, intermediate (VII)) with acid (XLII) (the acid derived from scheme 13922101a, intermediate ester (IX)) by means of 2-chloro-1,3-dimethylimidazolidinium hexafluorophosphate (CIP), and 1-hydroxy-7-azabenzotriazole (HOAt) in THF/dichloromethane gives the coupling product (XLIII), which is allylated with allyl bromide (XLIV) and Cs2CO3 in DMF yielding the allyl ether (XLV). The reduction of the lactone group of (XLV) with LiAlH2(OEt)2 in ethyl ether affords the lactol (XLVI), which is desilylated with KF in methanol to provide the phenolic compound (XLVII). The opening of the lactol ring of (XLVII) with simultaneous cyclization by means of Tf-OH in water/trifluoroethanol gives the hexacyclic intermediate (XLVIII), which is finally reductocondensed with KCN by means of LiAlH2(OEt)2 in THF to furnish the previously reported pentacyclic intermediate (XI) (scheme 13922101a, intermediate (XI)).
……………………………………………
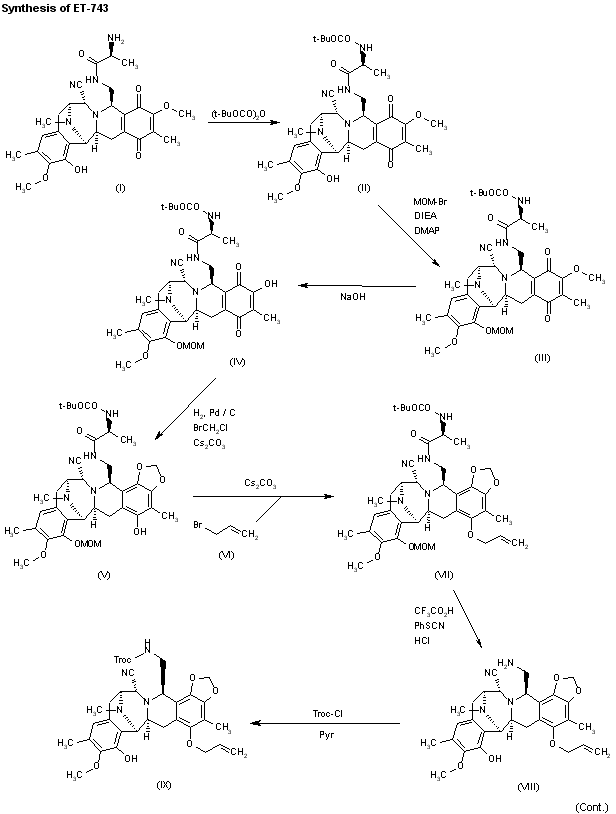
Reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino-protected compound (II), which is treated with methoxymethyl bromide (MOM-Br), DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF to provide compound (V). Reaction of (V) with allyl bromide (VI) and Cs2CO3 in DMF gives the allyl ether (VII), which first is treated with TFA, phenyl isothiocyanate and HCl to yield the primary amine (VIII) and then protected at the free NH2 group with Troc-Cl and pyridine, to afford the amino protected compound (IX).Org Lett 2000,2(16),2545
……………………………….
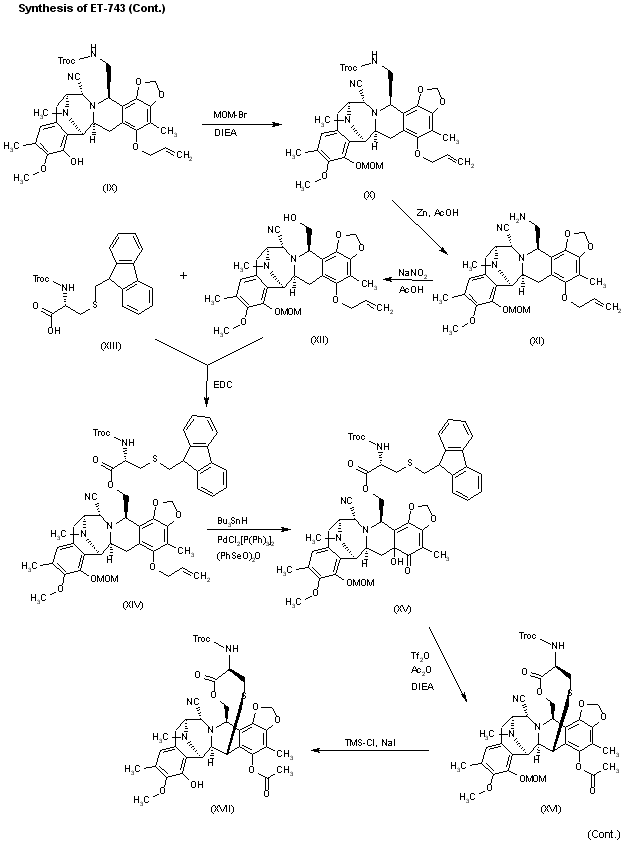
Reaction of (IX) with MOM-Br and DIEA as before affords the ether (X), which is treated with Zn/HOAc in order to regenerate the primary amino group giving (XI). The reaction of (XI) with NaNO2 and HOAc eliminates the NH2 group, affording the primary alcohol (XII), which is esterified with the protected (S)-cysteine (XIII) by means of EDC and DMAP in dichloromethane furnishing the cysteine ester (XIV). Reaction of (XIV) with Bu3SnH and PdCl2(PPh3)2, followed by oxidation with (PhSeO)2O in dichloromethane gives the hydroxyketone (XV), which is cyclized with Tf2O and Ac2O yielding the heptacyclic compound (XVI). Elimination of the MOM protecting group with TMSCl and NaI in CH3CN/CH2Cl2 affords the phenolic compound (XVII).
…………………….
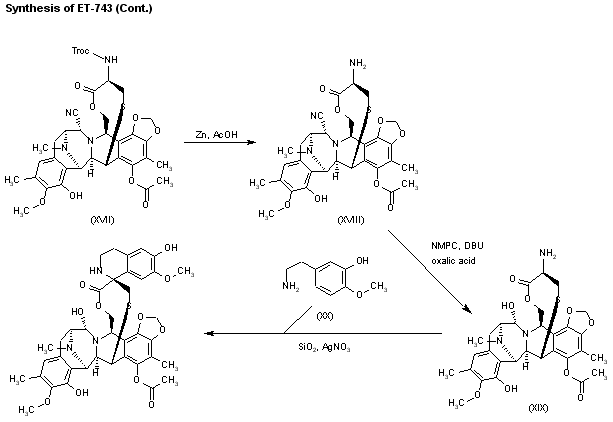
Intermediate (XVII) by a treatment with Zn and HOAc eliminates the Troc protecting group, giving the primary amine (XVIII). This compound by treatment with 4-formyl-1-methylpyridinium iodide (NMPC), DBU and oxalic acid in order to convert the nitrile group into an alcohol, provides compund (XIX), which is finally cyclized with 2-(3-hydroxy-4-methoxyphenyl)ethylamine (XX) by means of SiO2 / EtOH, followed treatment with and AgNO3 in acetonitrile/water.
……………………….
The reaction of cyanosafracin B (I) with Boc2O in ethanol gives the amino protected compound (II), which is treated with Mom-Br, DIEA and DMAP in acetonitrile yielding the O-protected compound (III). The demethylation of (III) with NaOH in methanol affords the hydroxyquinone (IV), which is reduced with H2 over Pd/C and cyclized with bromochloromethane and Cs2CO3 in hot DMF providing the methylenedioxy compound (V). The reaction of (V) with acetyl chloride and pyridine in dichloromethane gives the acetate (VI), which is treated with TFA, phenyl isothiocyanate and HCl yielding the primary amine (VII). Finally, this compound is treated with phthalic anhydride (VIII) and CDI in dichloromethane to afford the target phthalimide (phthalascidin Pt-650)
………………………………
Org. Lett., 2000, 2 (7), pp 993–996
DOI: 10.1021/ol0056729

Org. Lett., 2000, 2 (7), pp 993–996
DOI: 10.1021/ol0056729
…………………………
Enantioselective Total Synthesis of Ecteinascidin 743
Department of Chemistry, Harvard University Cambridge, Massachusetts 02138
J. Am. Chem. Soc., 1996, 118 (38), pp 9202–9203
DOI: 10.1021/ja962480t
……………………………….
Ecteinascidins are a group of marine alkaloid having antineoplasticity which is isolated from the extracted products from the marine tunicate habitat of the Caribbean sea by a very small amount. Arming the ecteinascidins, Et 743 has a very strong antineoplastic activity, studies to put it into practical use as a carcinostatic agent are limited, and the phase II clinical tests are now being carried out in ten countries in Europe and America. It is known that Et 743 has an effect of depressing the proliferation of cancer cells by 10 to 100 times more potent than (IC50=0.1-1 nM) Toxol, Camptotesin, Adriamycin or Mitomycin which are currently used carcinostatic agents.
From the background mentioned above, various studies for synthesis were carried out; however, the complete synthesis was only reported by Prof. E. J. Corey of Harvard University in the U.S.A. (J. Am. Chem. Soc. 1996, 118, 9202-9203, reference document A).
In the process of the total synthesis disclosed in Document A (refer to page 9202), the main feature of the process is that Et 743 is synthesized from the analogous compound to the compound represented by general formula 1 of the present invention via intermediates 4 and 8. That is, according to said process, the C4 site of ring B (regarding the location of rings, and the sites of atoms comprising the 6 membered ring, refer to general formula 1), which composes a 6 membered ring, is formed from the intermediate 4 at the first step. Since the atom C4 composing the ring B of the 6-membered ring H, which lacks reactivity, is bonded, it becomes necessary to perform an oxidation reaction at the C4 site on the B ring. This oxidation reaction is not effective and is carried out under harsh conditions; therefore production on an industrial scale is difficult, and also the yield is not good. Further, since the atom N12 site of the synthesized intermediate is substituted by an alkyl group which lacks reactivity, in this case substituted by a methyl group, it is not suited to the synthesis of various compounds. Although total synthesis was reported, the supplying source of Et 743 still depends on the natural sample whose supply is very scarce. Therefore, the establishment of the method for a large scale production of Et 743 is desired and requires accomplishing an effective synthesizing process.
Since ET 743 is known as a medicine having high antineoplasticity, and phthalascidin induced from the intermediate product at the synthesis of Et 743 displays the same activity to ET 743, the establishment of an effective and mild method for synthesis of ET 743 and analogous compounds thereof is strongly desired.
Therefore, the subject of the present invention is to accomplish the effective method for total synthesis of Et 743, and further, to provide not only Et 743 but also analogous compounds.
To dissolve the subject, the present invention uses retrosynthetic analysis for easy synthesis. It will be possible to form a B ring by a ring forming reaction at the ortho position of phenol, which binds an A ring to inner molecular aldehyde in a compound generated by the 4-8 reaction. Further, the present invention contemplates that the generated compound by the 4-8 reaction can be synthesized based on the polycondensation reaction of general formula 4, and general formula 5 via a compound of general formula 3. Then the total synthesis of Et 743, which is the aimed compound, can be accomplished by way of the compounds represented by general formulae 5, 4, 3, 2 and 1 and the specific structure of general formulae 1 and 2. This synthetic route provides for the analogous compounds of Et 743.





Mechanism of action
The biological mechanism of action is believed to involve the production of superoxide near the DNA strand, resulting in DNA backbone cleavage and cell apoptosis. The actual mechanism is not yet known, but is believed to proceed from reduction of molecular oxygen into superoxide via an unusual auto-redox reaction on a hydroxyquinone moiety of the compound following. There is also some speculation the compound becomes ‘activated’ into its reactive oxazolidine form.

Schematic of the unique and complex mode of action of trabectedin. The antitumor effects of trabectedin are due to multiple mechanisms involving DNA binding in the minor groove, interactions with DNA repair mechanisms, modulation of transcription regulation, and induction of microenvironment changes.
References
- Lichter et al. Worthen LW, ed. “Food-drugs from the sea. Proc: Aug 20–23, 1972.” 173. Marine Tech Soc. pp. 117–127.
- Rinehart KL (January 2000). “Antitumor compounds from tunicates”. Med Res Rev 20(1): 1–27. doi:10.1002/(SICI)1098-1128(200001)20:1<1::AID-MED1>3.0.CO;2-A.PMID 10608919.
- “Potent cancer drugs made — Sea squirts provide recipe”.
- Rath CM et al (November 2011). “Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743″. ACS Chemical Biology 6 (11): 1244–56. doi:10.1021/cb200244t.PMC 3220770. PMID 21875091.
- “New Scientist”.
- E. J. Corey, David Y. Gin, and Robert S. Kania (1996). “Enantioselective Total Synthesis of Ecteinascidin 743″. J. Am. Chem. Soc. 118 (38): 9202–9203.doi:10.1021/ja962480t.
- C. Cuevas et al. (2000). “Synthesis of ecteinascidin ET-743 and phthalascidin PT-650 from cyanosafracin”. B. Org. Lett. 2: 2545–2548.
- “CHMP evaluation”.
- “PharmaMar website”.
- S.Korea approves Zeltia cancer drug Yondelis, Reuters.com, May 8, 2008
- Grogan, Kevin (3 May 2011). “J&J pulls submission for Zeltia’s Yondelis”.PharmaTimes Magazine (London, England). Online PharmaTimes. Archived from the original on 7 May 2011. Retrieved 7 May 2011.
- “PharmaMar website”.
| |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (1′R,6R,6aR,7R,13S,14S,16R)-6′,8,14-trihydroxy-7′,9-dimethoxy-4,10,23-trimethyl-19-oxo-3′,4′,6,7,12,13,14,16-octahydrospiro[6,16-(epithiopropano-oxymethano)-7,13-imino-6aH-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1′(2′H)-isoquinolin]-5-yl acetate | |
| CLINICAL DATA | |
| AHFS/DRUGS.COM | International Drug Names |
| LICENCE DATA | EMA:Link |
| LEGAL STATUS | |
| ROUTES | Intravenous |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | Not applicable (IV only) |
| PROTEIN BINDING | 94 to 98% |
| METABOLISM | Hepatic (mostly CYP3A4-mediated) |
| HALF-LIFE | 180 hours (mean) |
| EXCRETION | Mostly fecal |
| IDENTIFIERS | |
| CAS NUMBER | 114899-77-3 |
| ATC CODE | L01CX01 |
| PUBCHEM | CID 108150 |
| IUPHAR LIGAND | 2774 |
| DRUGBANK | DB05109 |
| CHEMSPIDER | 16736970  |
| UNII | ID0YZQ2TCP |
| CHEMICAL DATA | |
| FORMULA | C39H43N3O11S |
| MOL. MASS | 761.84 g/mol |
……..
1 Corey, “Enantioselective Total Synthesis of Ecteinascidin 743“, J. Am. Chem. Soc. 1996, vol. 118, 9202-9203.
| 2 | * | Endo, “Synthetic Study on Ecteinascidin 743 Starting From D-Glucose“, Synlett 1999, No. 7, 1103-1105. |
| 3 | * | Endo, “Total Synthesis of Ecteinascidin 743“, J. Am. Chem. Soc. 2002, vol. 124, 6552-6554. |
| 4 | * | Hinterding, “Synthesis and In Vitro Evaluation of the Ras Farnesyltransferase Inhibitor Pepticinnamin E“, Angew. Chem. Int. Ed. 1998, 37, No. 9 1236-1239. |
| 5 | * | Tohma, “Synthesis of Optically Active alpha-Arylglycines: Stereoselective Mannich-Type Reaction with a New Chiral Template“, Synlett 2001, No. 7, 1179-1181.Hamprecht, D.W.; Berge, J.M.; Copley, R.C.B.; Eggleston, D.S.; Houge-Frydrych, C.S.V.; Jarvest, R.L.; Mensah, L.M.; O’Hanlon, P.J.; Pope, A.J.; Rittenhouse, S. Derivatives of the natural product SB-219383 and synthetic analogues: Potent inhibitors of bacterial tyrosyl tRNA synthetase 16th Int Symp Med Chem (September 18-22, Bologna) 2000, Abst PA-155Cuevas, C.; Perez, M.; Martin, M.J.; et al. Synthesis of ecteinascidin ET-743 and phathalascidin Pt-650 from cyanosafracin B Org Lett 2000, 2(16): 2545 |
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Assay for identifying biological targets of polynucleotide-binding compounds [US2008096201] | 2008-04-24 | |
| Compounds of the saframycin-ecteinascidin series, uses, and synthesis thereof [US6936714] | 2004-07-01 | 2005-08-30 |
| Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7807833] | 2009-08-06 | 2010-10-05 |
| Method For Total Synthesis Of Ecteinascidins And Intermediate Compounds Thereof [US7820838] | 2009-02-05 | 2010-10-26 |
| Assay for identifying biological targets of polynucleotide-binding compounds [US7183054] | 2004-12-09 | 2007-02-27 |
↧
↧
KEBUZONE…….An antirheumatic agent.
KEBUZONE…….An antirheumatic agent.

Kebuzone (or ketophenylbutazone) is a non-steroidal anti-inflammatory drug.
Structural formula

4-(3-oxobutyl)-1,2-diphenylpyrazolidine-3,5-dione
4-(3-Oxobutyl)-1,2-diphenyl-3,5-pyrazolidinedione
Additional Names: 1,2-diphenyl-4-(g-ketobutyl)-3,5-pyrazolidinedione; 1,2-diphenyl-4-(3¢-oxobutyl)-3,5-dioxopyrazolidine; ketophenylbutazone; KPB
Trademarks: Chebutan; Chepirol; Chetazolidin (Zeria); Chetil; Copirene; Ketason; Ketazone (Beytout); Pecnon (Sanken); Phloguron (Steiner); Recheton
MF: C19H18N2O3
MW: 322.36
Percent Comp: C 70.79%, H 5.63%, N 8.69%, O 14.89%
Properties: Crystals, mp 115.5-116.5° or 127.5-128.5° depending on cryst form.
Melting point: mp 115.5-116.5° or 127.5-128.5° depending on cryst form
Therap-Cat: Antirheumatic.
- BRN 0308507
- Chebutan
- Chepirol
- Chetazolidin
- Chetil
- Copirene
- EINECS 212-715-7
- Hichillos
- Kebuzone
- Kebuzonum
- Kebuzonum [INN-Latin]
- Keobutane-jade
- Ketason
- Ketazone
- Ketophenylbutazone
- Ketophenylbutazonum
- KPB
- Pecnon
- Quebuzona
- Quebuzona [INN-Spanish]
- Recheton
- UNII-4VD83UL6Y6
Anti-inflammatory agents that are non-steroidal in nature. In addition to anti-inflammatory actions, they have analgesic, antipyretic, and platelet-inhibitory actions.They act by blocking the synthesis of prostaglandins by inhibiting cyclooxygenase, which converts arachidonic acid to cyclic endoperoxides, precursors of prostaglandins. Inhibition of prostaglandin synthesis accounts for their analgesic, antipyretic, and platelet-inhibitory actions; other mechanisms may contribute to their anti-inflammatory effects.
UV – range
 | ||||
| Conditions : Concentration – 1 mg / 100 ml | ||||
| THE SOLVENT DESIGNATION GRAPHICS | METHANOL | WATER | 0.1М HCL | 0.1M NAOH |
|---|---|---|---|---|
| Maximum absorption | 244 nm | - | 237 nm | 262 nm |
 | 448 | - | 404 | 617 |
| e | 14440 | - | 13020 | 19890 |
IR – spectrum
| WAVELENGTH (ΜM) |
|---|
 |
| WAVE NUMBER (CM -1 ) |
Reference
- UV and IR Spectra. H.-W. Dibbern, R.M. Muller, E. Wirbitzki, 2002 ECV
- NIST/EPA/NIH Mass Spectral Library 2008
- Handbook of Organic Compounds. NIR, IR, Raman, and UV-Vis Spectra Featuring Polymers and Surfactants, Jr., Jerry Workman. Academic Press, 2000.
- Handbook of ultraviolet and visible absorption spectra of organic compounds, K. Hirayama. Plenum Press Data Division, 1967.
Brief background information
| SALT | ATC | FORMULA | MM | CAS |
|---|---|---|---|---|
| - | M01AA06 | C 19 H 18 N 2 O 3 | 322.36 g / mol | 853-34-9 |
| 4-(3-oxobutyl)-1,2-di(phenyl)pyrazolidine-3,5-dione | |
| CLINICAL DATA | |
|---|---|
| LEGAL STATUS | ? |
| IDENTIFIERS | |
| CAS NUMBER | 853-34-9 |
| ATC CODE | M01AA06 |
| PUBCHEM | CID 3824 |
| CHEMSPIDER | 3692  |
| UNII | 4VD83UL6Y6 |
| KEGG | D01567 |
| CHEBI | CHEBI:31749 |
| CHEMICAL DATA | |
| FORMULA | C19H18N2O3 |
| MOL. MASS | 322.35782 g/mol |
Application
- anti-inflammatory
- antirheumatic
- Synthesis pathway
| SYNTHESIS OF A) |
|---|
   |
| SYNTHESIS B) |
 |
Trade names
| COUNTRY | TRADE NAME | MANUFACTURER |
|---|---|---|
| Germany | Kebuzon | Steiner |
| France | Ketazon | Beytout |
| Italy | Chetopir | Sarm |
| Ukraine | no | no |
Formulations
- ampoules of 1 g / 5 ml;
- 250 mg capsule
Reference
- Synthesis of a)
- Denss, R. et al .: Helv. Chim. Acta (HCACAV) 40, 402 (1957).
- material:
- Kühn, M .: J. Prakt. Chem. (JPCEAO) 156 (II), 103 (1940).
- Synthesis b)
- AT 198 263 (Synfarma; appl. 1955).
References: Prepn: Deuss et al., US 2910481 (1959 to Geigy).
Review of pharmacology: Horakova et al.,Pharmacotherapeutica 1950-1959, 335-350 (1963), C.A. 60, 6072g (1964).
Metabolism: Nemecek et al., Arzneim.-Forsch. 16,1339 (1966); Queisnerova, Nemecek,Cesk. Farm. 20, 55 (1971), C.A. 75, 47077u (1971).
Herrenknecht, Christine; Guernet-Nivaud, Elisabeth; Lafont, Olivier; Guernet, Michel; Gueutin, Claire
Canadian Journal of Chemistry, 1988 , v. 66, pg. 1199 – 1202
Canadian Journal of Chemistry, 1988 , v. 66, pg. 1199 – 1202
Cizmarik; Lycka
Pharmazie, 1988 , v. 43, 11 pg. 794 – 795
Pharmazie, 1988 , v. 43, 11 pg. 794 – 795
Gueutin-Pelinard, Claire; Nivaud, Elisabeth; Boucly, Patrick; Guernet, Michel
Canadian Journal of Chemistry, 1981 , v. 59, pg. 759 – 762
Canadian Journal of Chemistry, 1981 , v. 59, pg. 759 – 762
Denss et al.
Helvetica Chimica Acta, 1957 , v. 40, pg. 402,406
Helvetica Chimica Acta, 1957 , v. 40, pg. 402,406
Patent: CS124279 , 1965 ;Chem.Abstr., 1968 , v. 69, 52134r
SPOFA; United Pharmaceutical Work Patent: FR1500627 , 1965 ;Chem.Abstr., 1968 , v. 69, 96715k
Nippon Shinyaju Co., Ltd. Patent: US5811547 A1, 1998 ;
Fisnerova,L. et al. Collection of Czechoslovak Chemical Communications, 1974 , v. 39, pg. 624 – 633
↧
EU approves Lilly diabetes drug Trulicity, dulaglutide
EU approves Lilly diabetes drug Trulicity, dulaglutide
Regulators in Europe have given the green light to Eli Lilly’s Trulicity, its once-weekly glucagon-like peptide-1 receptor agonist for type 2 diabetes.
Read more at: http://www.pharmatimes.com/Article/14-11-25/EU_approves_Lilly_diabetes_drug_Trulicity.aspx
Dulaglutide is a glucagon-like peptide 1 receptor agonist (GLP-1 agonist) for the treatment of type 2 diabetes that can be used once weekly.[1][2]GLP-1 is a hormone that is involved in the normalization of level of glucose in blood (glycemia). The FDA approved dulaglutide for use in the United States in September 2014.[3] The drug is manufactured by Eli Lilly under the brand name Trulicity.[3]

Mechanism of action
Dulaglutide binding to glucagon-like peptide 1 receptor, slows gastric emptying and increases insulin secretion by beta cells in the pancreas. Simultaneously the compound reduces the elevated glucagon secretion by alpha cells of the pancreas, which is known to be inappropriate in the diabetic patient. GLP-1 is normally secreted by L cells of the gastrointestinal mucosa in response to a meal.[4]
Medical uses[
The compound is indicated for adults with type 2 diabetes mellitus as an adjunct to diet and exercise to improve glycemic control. Dulaglutide is not indicated in the treatment of subjects with type 1 diabetes mellitus or patients with diabetic ketoacidosis. Dulaglutide can be used either stand-alone or in combination with other medicines for type 2 diabetes, in particularmetformin, sulfonylureas, thiazolidinediones, and insulin taken concomitantly with meals.[5]
Side effects
The most common side effects include gastrointestinal disorders, such as dyspepsia,decreased appetite, nausea, vomiting, abdominal pain, diarrhea.[6] Some patients may experience serious adverse reactions: acute pancreatitis (symptoms include persistent severe abdominal pain, sometimes radiating to the back and accompanied by vomiting),hypoglycemia, renal impairment (which may sometimes require hemodialysis). The risk of hypoglycemia is increased if the drug is used in combination with sulfonylureasorinsulin.[7][8]
Contraindications
The compound is contraindicated in subjects with hypersensitivity to active principle or any of the product’s components. As a precautionary measure patients with a personal or family history of medullary thyroid carcinoma or affected by multiple endocrine neoplasia syndrometype 2 should not take dulaglutide, because for now it is unclear whether the compound can increase the risk of these cancers.[9]
References
- JCourtney Aavang Tibble, Tricia Santos Cavaiola, Robert R Henry (2013). “Longer Acting GLP-1 Receptor Agonists and the Potential for Improved Cardiovascular Outcomes: A Review of Current Literature”. Expert Rev Endocrinol Metab 8 (3): 247–259.doi:10.1586/eem.13.20.
- “Lilly’s Once-Weekly Dulaglutide Shows Non-Inferiority to Liraglutide in Head-to-Head Phase III Trial for Type 2 Diabetes”. Eli Lilly. Feb 25, 2014.
- “FDA approves Trulicity to treat type 2 diabetes” (Press release). FDA. Sep 18, 2014.
- Nadkarni P, Chepurny OG, Holz GG (2014). “Regulation of glucose homeostasis by GLP-1″. Prog Mol Biol Transl Sci 121: 23–65. doi:10.1016/B978-0-12-800101-1.00002-8.PMC 4159612. PMID 24373234. Retrieved 2014-09-29.
- Terauchi Y, Satoi Y, Takeuchi M, Imaoka T (July 2014). “Monotherapy with the once weekly GLP-1 receptor agonist dulaglutide for 12 weeks in Japanese patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomised, double-blind, placebo-controlled study”. Endocr. J. PMID 25029955. Retrieved 2014-09-29.
- Nauck M, Weinstock RS, Umpierrez GE, Guerci B, Skrivanek Z, Milicevic Z (August 2014). “Efficacy and safety of dulaglutide versus sitagliptin after 52 weeks in type 2 diabetes in a randomized controlled trial (AWARD-5)”. Diabetes Care 37 (8): 2149–58.doi:10.2337/dc13-2761. PMID 24742660.
- Amblee A (April 2014). “Dulaglutide for the treatment of type 2 diabetes”. Drugs Today50 (4): 277–89. doi:10.1358/dot.2014.50.4.2132740. PMID 24918645.
- Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E (February 2014). “Glucagon-like peptide-1 receptor agonists and pancreatitis: a meta-analysis of randomized clinical trials”. Diabetes Res. Clin. Pract. 103 (2): 269–75.doi:10.1016/j.diabres.2014.01.010.PMID 24485345.
- Samson SL, Garber A (April 2013). “GLP-1R agonist therapy for diabetes: benefits and potential risks”. Curr Opin Endocrinol Diabetes Obes 20 (2): 87–97.doi:10.1097/MED.0b013e32835edb32. PMID 23403741. Retrieved 2014-09-30.
| IDENTIFIERS | |
|---|---|
| CAS NUMBER | 923950-08-7 |
| ATC CODE | None |
| CHEMICAL DATA | |
| FORMULA | C2646H4044N704O836S18 |
| MOL. MASS | 59669.81 g/mol |
↧
Antibacterial activities and antioxidant capacity of Aloe vera
Antibacterial activities and antioxidant capacity of Aloe vera
Organic and Medicinal Chemistry Letters 2013, 3:5 doi:10.1186/2191-2858-3-5
The electronic version of this article is the complete one and can be found online at:http://www.orgmedchemlett.com/content/3/1/5
Background
The aim of this study was to identify, quantify, and compare the phytochemical contents, antioxidant capacities, and antibacterial activities of Aloe vera lyophilized leaf gel (LGE) and 95% ethanol leaf gel extracts (ELGE) using GC-MS and spectrophotometric methods.
↧
↧
Peptide Has Promise for Treating Spinal Cord Injury
FEATURED STORY Peptide Has Promise for Treating Spinal Cord Injuryread athttp://www.dddmag.com/news/2014/12/peptide-has-promise-treating-spinal-cord-injury?et_cid=4300209&et_rid=523035093&type=cta | |||||||||
Case Western Reserve scientists have developed a new chemical compound that shows extraordinary promise in restoring function lost to spinal cord injury. The compound, which the researchers dubbed intracellular sigma peptide (ISP), allowed paralyzed muscles to activate in more than 80 percent of the animals tested. Read more...
|


↧
LEXIPAFANT
Lexipafant
CAS : 139133-26-9
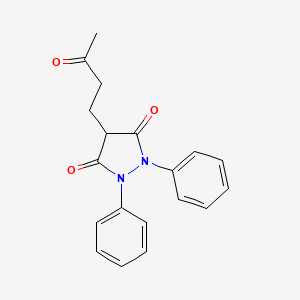
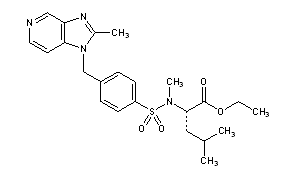
N-Methyl-N-[[4-[(2-methyl-1H-imidazo[4,5-c]pyridin-1-yl)methyl]phenyl]sulfonyl]-L-leucine ethyl ester
N-methyl-N-[[a-(2-methyl-1H-imidazo[4,5-c]pyridin-1-yl)-p-tolyl]sulfonyl]-L-leucine ethyl ester
N-Methyl-N-[4-(2-methyl-1H-imidazo[4,5-c]pyridin-1-ylmethyl)phenylsulfonyl]-L-leucine ethyl ester
Manufacturers’ Codes: BB-882
DO6
GR-167089
ISV-611
GR-167089
ISV-611
UNII-H14917M9YW
Trademarks: Zacutex (Brit. Biotech)
MF: C23H30N4O4S
M Wt: 458.57
Percent Composition: C 60.24%, H 6.59%, N 12.22%, O 13.96%, S 6.99%
Properties: White crystalline solid from ethyl acetate, mp 105°. [a]D20 -6.7° (c = 2.0 in CDCl3).
Melting point: mp 105°
Optical Rotation: [a]D20 -6.7° (c = 2.0 in CDCl3)
Therap-Cat: Anti-inflammatory. (Nonsteroidal); Platelet Activating Factor Antagonist.
Lexipafant is a platelet-activating factor (PAF) antagonist that was in early clinical development at DevCo for the oral treatment of dementia and motor function disorders in HIV patients, intravenous treatment of acute pancreatitis, as well as for the prevention of certain serious renal and neurological complications experienced by patients undergoing cardiac surgery, including stroke. However, no recent developments of the drug candidate have been reported by the company.
Lexipafant was also being studied at British Biotech (now Vernalis) for the intravenous treatment of pancreatitis, but development for this indication was discontinued. In 2002, DevCo obtained from British Biotech exclusive rights to develop, manufacture and sell lexipafant for the treatment of human disease, excluding the fields of oncology and ophthalmology.
……………………………
……………………………………………
WO 1993016075
………………………………
WO 1995013064
Literature References:
Platelet activating factor (PAF) antagonist. Prepn: M. Whittaker, A. Miller, WO 9203422; eidem, US5200412 (1992, 1993 both to British Bio-Technology).
Structure-activity report: M. Whittaker et al., J. Lipid Mediators Cell Signalling 10, 151 (1994).
Pharmacology: F. M. Abu-Zidan et al., Pharmacol. Toxicol. 78, 23 (1996).
Clinical evaluation in acute pancreatitis: A. N. Kingsnorth et al., Br. J. Surg. 82, 1414 (1995).





![]() amcrasto@gmail.com
amcrasto@gmail.com

↧
20 Superb Herbal Remedies for Abdominal Fat

read all
http://www.rapidhomeremedies.com/remedies-for-abdominal-fat.html
boost your metabolism excellently for the whole day.
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL ![]()

![]() amcrasto@gmail.com
amcrasto@gmail.com
↧
KAE 609, NITD 609, Cipargamin
 Cipargamin, NITD 609
Cipargamin, NITD 609IUPAC Name: (3R,3'S)-5,7'-dichloro-6'-fluoro-3'-methylspiro[1H-indole-3,1'-2,3,4,9-tetrahydropyrido[3,4-b]indole]-2-one |
CAS Registry Number: 1193314-23-6
Synonyms: NITD609, NITD 609, NITD-609, GNF-609
KAE-609
NITD-609
Synonyms: NITD609, NITD 609, NITD-609, GNF-609
KAE-609
NITD-609
390.238, C19 H14 Cl2 F N3 O
(1'R,3'S)-5,7'-Dichloro-6'-fluoro-3'-methyl-1,2,2',3',4',9'-hexahydrospiro[indole-3,1'-pyrido[3,4-b]indole]-2-one
(1R,3S)-5′,7-Dichloro-6-fluoro-3-methyl-2,3,4,9-tetrahydrospiro[β-carboline-1,3′-indol]-2′(1′H)-one

NITD609 is an experimental synthetic antimalarial molecule belonging to the spiroindolone class.[1][2] The compound was developed at the Novartis Institute for Tropical Diseases in Singapore, through a collaboration with the Genomics Institute of the Novartis Research Foundation (GNF), the Biomedical Primate Research Centre and the Swiss Tropical Institute. NITD609 is a novel, synthetic antimalarial molecule belonging to the spiroindolone class, awarded MMV Project of the Year 2009.
It is structurally related to GNF 493, a compound first identified as a potent inhibitor of Plasmodium falciparum growth in a high throughput phenotypic screen of natural products conducted at the Genomics Institute of the Novartis Research Foundation in San Diego, California in 2006. NITD609 was discovered by screening the Novartis library of 12,000 natural products and synthetic compounds to find compounds active against Plasmodium falciparum. The first screen turned up 275 compounds and the list was narrowed to 17 potential candidates.
KAE609 (cipargamin; formerly NITD609, Novartis Institute for Tropical Diseases) is a new synthetic antimalarial spiroindolone analogue with potent, dose-dependent antimalarial activity against asexual and sexual stages of Plasmodium falciparum.http://www.nejm.org/doi/full/10.1056/NEJMoa1315860
KAE609 shows promise as next generation treatment for malaria
http://www.novartis.com/newsroom/media-releases/en/2014/1843976.shtml- KAE609 is the first antimalarial drug candidate with a novel mechanism of action to achieve positive clinical proof-of-concept in over 20 years
- KAE609 was tested in adult patients with uncomplicated malaria and showed a median parasite clearance time of 12 hours, including in patients with resistant infections[1]
- For more than a decade, Novartis has been a leader in the fight against malaria, setting the current gold standard for treatment and building one of the strongest malaria pipelines in the industry

KAE609 shows promise as next generation treatment for malaria
- KAE609 is the first antimalarial drug candidate with a novel mechanism of action to achieve positive clinical proof-of-concept in over 20 years
- KAE609 was tested in adult patients with uncomplicated malaria and showed a median parasite clearance time of 12 hours, including in patients with resistant infections[1]
- For more than a decade, Novartis has been a leader in the fight against malaria, setting the current gold standard for treatment and building one of the strongest malaria pipelines in the industry
The digital press release with multimedia content can be accessed here:
![]()

Basel, Switzerland, July 30, 2014- Today, Novartis published clinical trial results in the New England Journal of Medicine showing that KAE609 (cipargamin), a novel and potent antimalarial drug candidate, cleared the parasite rapidly in Plasmodium falciparum (P. falciparum) and Plasmodium vivax (P. vivax) uncomplicated malaria patients[1]. Novartis currently has two drug candidates in development. Both KAE609 and KAF156 are new classes of anti-malarial compounds that treat malaria in different ways from current therapies, important to combat emerging drug resistance. Novartis has also identified PI4K as a new drug target with potential to prevent, block and treat malaria.
"Novartis is in the fight against malaria for the long term and we are committed to the continued research and development of new therapies to eventually eliminate the disease," said Joseph Jimenez, CEO of Novartis. "With two compounds and a new drug target currently under investigation, Novartis has one of the strongest malaria pipelines in the industry."
Malaria is a life-threatening disease primarily caused by parasites (P. falciparum and P. vivax) transmitted to people through the bites of infected Anopheles mosquitoes. Each year it kills more than 600,000 people, most of them African children[2].
"KAE609 is a potential game-changing therapy in the fight against malaria," said Thierry Diagana, Head of the Novartis Institute for Tropical Diseases (NITD), which aims to discover novel treatments and prevention methods for major tropical diseases. "Novartis has given KAE609 priority project status because of its unique potential of administering it as a single-dose combination therapy."
In June 2012, 21 patients infected by one of the two main malaria-causing parasite types took part in a proof-of-concept clinical study conducted in Bangkok and Mae Sot near the Thailand/Burma border where resistance to current therapies had been reported. Researchers saw rapid parasite clearance in adult patients (median of 12 hours)[2] with uncomplicated P. vivax or P. falciparum malaria infection including those with resistant parasites. No safety concerns were identified, however the study was too small for any safety conclusions.
"The growing menace of artemisinin resistance threatens our current antimalarial treatments, and therefore our attempts to control and eliminate falciparum malaria," said Nick White, Professor of Tropical Medicine at Mahidol University in Thailand and lead author of the NEJM article. "This is why we are so enthusiastic about KAE609; it is the first new antimalarial drug candidate with a completely novel mechanism of action to reach Phase 2 clinical development in over 20 years."
KAE609, the first compound in the spiroindolone class of treatment, works through a novel mechanism of action that involves inhibition of a P-type cation-transporter ATPase4 (PfATP4), which regulates sodium concentration in the parasite. Because KAE609 also appears to be effective against the sexual forms of the parasite, it could potentially help prevent disease transmission. The clinical trial was done in collaboration with the Wellcome Trust-Mahidol University - Oxford Tropical Medicine Research Programme. Research was supported by the Wellcome Trust, Singapore Economic Development Board, and Medicines for Malaria Venture.
KAE609 represents one of two new classes of antimalarial compounds that Novartis has discovered and published in the last four years.[3],[4] This drug candidate has shown potent in vitro activity against a broad range of parasites that have developed drug resistance against current therapies. KAE609 is currently being planned for Phase 2b trials.
References
[1] http://www.nejm.org/doi/full/10.1056/NEJMoa1315860
[2] World Health Organization, http://www.who.int/mediacentre/factsheets/fs094/en/
[3] Spiroindolones, a Potent Compound Class for the Treatment of Malaria, KAE609, Science, Sept. 2010
[4] Imaging of Plasmodium liver stages to drive next generation antimalarial drug discovery. Science Express, Nov. 17, 2011
[1] http://www.nejm.org/doi/full/10.1056/NEJMoa1315860
[2] World Health Organization, http://www.who.int/mediacentre/factsheets/fs094/en/
[3] Spiroindolones, a Potent Compound Class for the Treatment of Malaria, KAE609, Science, Sept. 2010
[4] Imaging of Plasmodium liver stages to drive next generation antimalarial drug discovery. Science Express, Nov. 17, 2011

The current spiroindolone was optimized to address its metabolic liabilities leading to improved stability and exposure levels in animals. As a result, NITD609 is one of only a handful of molecules capable of completely curing mice infected withPlasmodium berghei (a model of blood-stage malaria).
Given its good physicochemical properties, promising pharmacokinetic and efficacy profile, the molecule was recently approved as a preclinical candidate and is now entering GLP toxicology studies with the aim of entering Phase I studies in humans in late 2010. If its safety and tolerability are acceptable, NITD609 would be the first antimalarial not belonging to either the artemisinin or peroxide class to go into a proof-of-concept study in malaria.
If NITD609 behaves similarly in people to the way it works in mice, it may be possible to develop it into a drug that could be taken just once - far easier than current standard treatments in which malaria drugs are taken between one and four times a day for up to seven days. NITD609 also has properties which could enable it to be manufactured in pill form and in large quantities. Further animal studies have been performed and researchers have begun human-stage trials.
| |
| Identifiers | |
| ChemSpider | 24662493 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C19H14Cl2FN3O |
| Molar mass | 390.24 g mol−1 |
Leishmaniasis is caused by one of more than twenty (20) varieties of parasitic protozoa that belong to the genus Leishmania, and is transmitted by the bite of female sandflies. Leishmaniasis is endemic in some 90 countries, including many tropical and sub-tropical areas.
There are four main forms of leishmaniasis. Visceral leishmaniasis, also called kala-azar, is the most serious form and is caused by the parasite Leishmania donovani. Patients who develop visceral leishmaniasis can die within months unless they receive treatment. The two main therapies for visceral leishmaniasis are the antimony derivatives sodium stibogluconate (Pentostam®) and meglumine antimoniate (Glucantim®). Sodium stibogluconate has been used for about 70 years and resistance to this drug is a growing problem. In addition, the treatment is relatively long and painful, and can cause undesirable side effects. Human African Trypanosomiasis, also known as sleeping sickness, is a vector-bome parasitic disease. The parasites concerned are protozoa belonging to the Trypanosoma Genus. They are transmitted to humans by tsetse fly {Glossina Genus) bites which have acquired their infection from human beings or from animals harbouring the human pathogenic parasites.
Chagas disease (also called American trypanosomiasis) is another human parasitic disease that is endemic amongst poor populations on the American continent. The disease is caused by the protozoan parasite Trypanosoma cruzi, which is transmitted to humans by blood-sucking insects. The human disease occurs in two stages: the acute stage, which occurs shortly after the infection, and the chronic stage, which can develop over many years. Chronic infections result in various neurological disorders, including dementia, damage to the heart muscle and sometimes dilation of the digestive tract, as well as weight loss. Untreated, the chronic disease is often fatal.
The drugs currently available for treating Chagas disease are nifurtimox and benznidazole. However, problems with these current therapies include their adverse side effects, the length of treatment, and the requirement for medical supervision during treatment. Furthermore, treatment is really only effective when given during the acute stage of the disease. Resistance to the two frontline drugs has already arisen. The antifungal agent amphotericin b has been proposed as a second-line drug, but this drug is costly and relatively toxic.
SYNTHESIS..........WILL BE UPDATED
...........................................
 http://www.google.com/patents/WO2009132921A1?cl=en
http://www.google.com/patents/WO2009132921A1?cl=enSCHEME G: Preparation of (lR,3S)-5',7-dichloro-6-fluoro-3-methyl-2,3,4,9- tetrahydrospiro[β-carboline-l,3'-indol-2'(l'iϊ)-one (35) and (lR,3S)-5'-chloro-6-fluoro-3- methyl-2,3,4,9-tetrahydrospiro[β-carboline-l,3'-indoI-2'(l'H0-one (36)

Step 2: The solution (0.2 M) of 6-chloro-5-fluoro-1H-indole-3-carbaldehyde (4.0 g, 20.24 mmol) in nitroethane (100 mL) was refluxed with ammonium acetate (1.32 g, 0.85 mmol) for 4 hours. The reaction mixture was concentrated under vacuum to remove nitroethane, diluted with ethylacetate and washed with brine. The organic layer was concentrated to give 6-chloro-5- fluoro-3-(2-nitro-propenyl)-1H-indole (5.0 g, 97 %) as a reddish orange solid. 1H ΝMR (500 MHz, CDCl3): δ 8.77 (s, IH), 8.32 (s, IH), 7.58 (d, IH, J= 2.5 Hz), 7.54 (d, IH, J = 9 Hz), 7.50 (d, IH, J= 5.9 Hz), 2.52 (s, 3H). Step 3: A solution of 6-chloro-5-fluoro-3-(2-nitro-propenyl)-1H-indole (5.0 g, 19.63 mmol) in tetrahydrofuran (10 mL) was added to the suspension of lithium aluminium hydride (2.92 g, 78.54 mmol) in tetrahydrofuran (20 mL) at 0 0C and then refluxed for 3 hours. The reaction mixture was cooled to 0 °C, and quenched according to the Fischer method. The reaction mixture was filtered through celite and the filtrate concentrated to give 2-(6-chloro-5-fluoro-1H-indol-3- yl-1-methyl-ethylamine (4.7 g crude) as a viscous brown liquid. The residue was used without further purification. 1H NMR (500 MHz, CDCl3): δ 8.13 (s, IH), 7.37 (d, IH, 6.Hz), 7.32 (d, IH, J = 10 Hz), 7.08 (s, IH), 3.23-3.26 (m, IH), 2.77-2.81 (m, IH), 2.58-2.63 (m, IH), 1.15 (d, 3H, J= 6.5 Hz).
Step 4: A mixture of 2-(6-chloro-5-fluoro-1H-indol-3-yl-l-methyl-ethylamine (4.7 g, 20.73 mmol), 5-chloroisatin (3.76 g, 20.73 mmol) and p-toluenesulphonic acid (394 mg, 2.07 mmol) in ethanol (75 mL) was refluxed overnight. The reaction mixture was concentrated to remove ethanol, diluted with ethyl acetate and washed with saturated aqueous NaHCO3. The organic layer was concentrated to give a brown residue, which was purified by silica gel chromatography (20 % ethyl acetate in hexane) to provide the corresponding racemate (4.5 g, 56 %) as a light yellow solid. The racemate was separated into its enantiomers by chiral chromatography to provide 35.
Compound 36 can be obtained in a similar fashion from 5-fluoroindole.
Alternatively 35 and 36 were be prepared in enantiomerically pure form by the following scheme.
SCHEME H: Alternative preparation of (lR,3S)-5',7-dichloro-6-fluoro-3-methyl-2,3,4,9- tetrahydrospiro[β-carboline-l,3'-indol-2'(1'H)-one (35)

Step 2: 2-Acetylamino-3-(6-chloro-5-fluoro-1H-indol-3-yl)-proprionic acid (2.5g, 8.4mmol) was dissolved in aqueous NaOH (IN, 10 niL) and water added (70 mL). The mixture was heated to 37-380C and neutralized with HCl (IN) to pΗ 7.3-7.8. L-Aminoacylase (0.5 g) was added to the mixture and allowed to stir for 2 days, maintaining 37-380C and pΗ 7.3-7.8. The mixture was heated to 60 °C for another hour, concentrated to remove part of water, cooled and filtered. The filtrate was adjusted to pΗ 5.89 and filtered again. The filtrate was adjusted to pΗ 2.0 and extracted with EtOAc. The combined organic layer was dried over Na2SO4, filtered, concentrated and the residue was purified with chromatography (petroleum ether /EtOAc 1 : IEtOAc) to give R- 2-acetylamino-3-(6-chloro-5-fluoro-1H-mdol-3-yl)-propπonic acid as a light yellow solid (1.2 g, 48% yield). Step 3: R-2-acetylamino-3-(6-chloro-5-fluoro-1H-indol-3-yl)-proprionic acid (1.2 g, 4.0 mmol) was dissolved in HCl (6N, 10 mL) and the mixture heated to reflux for 4 hours, and then concentrated to dryness. Toluene (50 mL) was added to the residue and concentrated to dryness to remove water and HCl. The residue was dried under vacuum and then dissolved in MeOH (20 mL). To the solution was added dropwise SOCl2 (0.5 mL, 6.8 mmol) at 0 °C, and the mixture was stirred overnight. After removal of solvent, the residue was dissolved in THF/water (40/10 mL) and NaHCO3 (1.0 g, 11.9 mmol) was added portionwise. Upon basifϊcation, BoC2O (1.2 g, 5.5 mmol) added at 0 °C and allowed to stir at room temperature. After TLC indicated the reaction was finished, EtOAc was added and separated and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with water and brine, dried with Na2SO4, filtered, concentrated and the residue was purified with chromatography (petroleum ether /EtOAc: 5/1) to give R-2-tert-butoxycarbonylamino-3-(6-chloro-5-fluoro-l/-/-indol-3-yl)-proprionic acid methyl ester 460 g, 31% yield for 3 steps).
Step 4: To a solution of R-2-tert-butoxycarbonylamino-3-(6-chloro-5-fluoro-l//-indol-3-yl)- proprionic acid methyl ester (460mg, 1.2mmol) in dry ether (20 mL) was added portionwise LiAlH4 (92 mg, 2.4 mmol) at 0 °C. The mixture was heated to reflux for 2 hours. After TLC indicated the reaction was finished, the mixture was cooled and carefully quenched with Na2SO4. The mixture was filtered and the filtrate was washed with saturated aqueous NH4Cl and water, dried with Na2SO4, filtered, concentrated to give a crude product (400 mg), which was used without further purification.
Step 5: To a solution of the crude product (400 mg, 1.2mmol) and Et3N (0.3 mL, 2.2 mmol) in CH2Cl2 (5 mL) was added MsCl (160 mg, 1.4 mmol) dropwise at 0 °C. The mixture was stirred for 2 hours at room temperature. After TLC indicated the reaction was completed, the mixture was washed with water and brine, dried with Na2SO4, filtered, concentrated and the residue was purified with chromatography (petroleum ether/EtOAc 5:1) to give methansulfonic acid (R)-2- ?ert-butoxycarbonylamino-3-(6-chloro-5-fluoro-1H-indol-3-yl)-propyl ester as a light yellow solid (300 mg, 57% yield, 2 steps)
Step 6: To a solution of mesylate (300 mg, 0.7mmol) in dry ether (20 mL) was added portionwise LiAlH4 (55 mg, 1.4 mmol) at 0 °C. The mixture was stirred at room temperature overnight. After TLC indicated the reaction was finished, the mixture was cooled and carefully quenched with Na2SO4. The mixture was filtered and the filtrate was washed with saturated aqueous NH4Cl and water, dried with Na2SO4, filtered, concentrated and the residue was purified with chromatography (petroleum ether/EtOAc 10: 1) to give [(5)-2-(6-chloro-5-fluoro-1H-indol-3-yl)- 1 -methyl-ethyl] -carbamic acid tert-butyl ester as a light yellow solid (200 mg, 87% yield).
Step 7: A solution of [(S)-2-(6-chloro-5-fluoro-1H-indol-3-yl)-l-methyl-ethyl]-carbamic acid tert-butyl ester (200 mg, 0.6 mmol) in HCl/MeOH (10 mL) was stirred at room temperature. After TLC indicated the reaction was finished, the mixture was concentrated to remove the solvent. To the residue was added EtOAc (5OmL), and the mixture was neutralized with saturated NaHCO3 to pH 8~9, and then extracted with EtOAc. The combined organic phases were dried with Na2SO4, filtered, concentrated to give a crude (S)-2-(6-chloro-5-fluoro-1H-indol-3-yl)-l- methyl-ethylamine which was used without further purification.
Step 8: To a solution of (5)-2-(6-chloro-5-fluoro-1H-indol-3-yl)-l-methyl-ethylamine (120 mg, 0.5 mmol) in EtOH (1OmL) was added 5-chloroisatin (90 mg, 0.5 mmol) and p-TsOΗ (8 mg, 0.04 mmol). The mixture was heated in a sealed tube at 1100C for 16 hours. After TLC indicated the reaction was finished, the mixture was cooled and concentrated. The residue was dissolved in EtOAc (2OmL) and washed with NaOH (IN) and brine, dried with Na2SO4, filtered, concentrated and the residue was purified with chromatography (petroleum ether/EtOAc 5:1) to give 36 (150mg, 64% yield over two steps).
Example 48 (15,3R)-5'-Chloro-3-methyl-2,3,4,9-tetrahydrospiro[β-carboline-l,3'-indol]-2'(l'JH)-one
(35)

Compound 35 may be prepared according to Scheme F using the same or analogous synthetic techniques and/or substituting with alternative reagents.
(lS^RVS'-Chloro-S-methyl-l^^^-tetrahydrospirotβ-carboline-l.S'-indoll-l^l'ZO-one: 1H NMR (300 MHz, DMSO-^6): δ 10.45 (s, IH), 10.42 (s, IH), 7.43 (d, J= 7.5 Hz, IH), 7.31 (dd, J = 2.1, 8.4 Hz, IH), 7.16 (d, J = 7.2 Hz, IH), 7.05-7.02 (m, 2H), 7.00-6.96 (m, IH), 6.92 (d, J = 8.1 Hz, IH), 3.98-3.86 (m, IH), 2.78 (dd, J= 3.6, 14.9 Hz, IH), 2.41 (dd, J= 4.5, 25.5 Hz, IH), 1.18 (d, J= 6.3 Hz, 3H); MS (ESI) m/z 338.0 (M+H)+.
Chiral compounds such as 36 and 37 can be prepared according to Scheme G or H using the same or analogous synthetic techniques and/or substituting with alternative reagents. Example 49
(IR^^-S'.T-Dichloro-ό-fluoro-S-methyl-l^^^-tetrahydrospiroIβ-carboline-l^'-indol]- 2\VH)-one (36)

35: 1H NMR (500 MHz, DMSO-Jd) δ 10.69 (s, IH), 10.51 (s, IH), 7.43 (d, J = 10.0 Hz, IH), 7.33 (dd, J= 8.4, 2.2 Hz, IH), 7.27 (d, J= 6.5 Hz, IH), 7.05 (d, J= 2.3, IH), 6.93 (d, J= 8.5 Hz, IH), 3.91 (m, IH), 3.13 (bd, J= 6.2 Hz, IH), 2.74 (dd, J= 15.0 , 3.0 Hz, IH), 2.35 (dd, J= 15.0, 10.3, IH), 1.15 (d, J= 6.0, 3H);
MS (ESI) m/z 392.0 (M+2H)+;
[α]25D = + 255.4°
Example 50
(lS,3R)-5',7-Dichloro-6-fluoro-3-methyI-2,3,4,9-tetrahydrospiro[β-carboline-l,3'-indol]- 2'(l'H)-one (37)

(lS^^-S'^-Dichloro-o-fluoro-S-methyl^jS^^-tetrahydrospirojP-carboline-l-S'-indol]- 2'(l'H)-one: 1H NMR (500 MHz, CDCl3) δ 8.49 (s, IH), 7.54 (s, IH), 7.24 (d, J= 9.7 Hz, IH), 7.21 (dd, J = 8.6, 2.0 Hz, IH), 7.14 (d, J= 6.0 Hz, IH), 7.11 (d, J= 1.8, IH), 6.77 (d, J= 8.3 Hz, IH), 4.14 (m, IH), 2.89 (dd, J = 15.4, 3.7 Hz, IH), 2.49 (dd, J = 15.3, 10.5, IH), 1.68 (bs, IH), 1.29 (d, J= 6.4 Hz, 3H); MS (ESI) m/z 392.0 (M+2H)+; [α]25D -223.3°
.............................................
US 2011275613
http://www.google.com/patents/WO2013139987A1?cl=enPrior art:
(1 'R, 3'S)-5, 7'-dichloro-6'-fIuoro-3'-methyl-2', 3',4', 9'-tetrahydrospiro[indoline-3, 1 - pyrido[3,4-b]indol]-2-one (eg. a compound of formula (IV), which comprises a spiroindolone moiety) and a 6-steps synthetic method for preparing, including known chiral amine intermediate compound (MA) are known (WO 2009/132921 ):

he present invention relates to processes for the preparation of spiroindolone compounds, such as (1'R,3'S)-5, 7'-dichloro-6'-fIuoro-3'-methyl-2',3',4',9'- tetrahydrospiro[indoline-3, 1 '-pyhdo[3.4-b]indol]-2-one.
(1 'R, 3'S)-5, 7'-dichloro-6'-fluoro-3'-methyl-2', 3',4 9'-tetrahydrospiro[indoline-3, 1 '- pyrido[3, 4-b]indol]-2-one is useful in the treatment and/or prevention of infections such as those caused by Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, Trypanosoma cruzi and parasites of the Leishmania genus such as, for example, Leishmania donovani., and it has the following structure:
![Figure imgf000009_0001]() (IVA)
(IVA)
(1 'R, 3'S)-5, 7'-dichloro-6'-fluoro-3'-methyl-2 3', 4', 9'-tetrahydrospiro[indoline-3, 1 - pyhdo[3, 4-b]indol]-2-one and a synthesis thereof are described in WO 2009/132921 Al in particular in Example 49 therein.
![Figure imgf000024_0002]()
(1 'R, 3'S)-5, 7'-dichloro-6'-fluoro-3'-methyl-2', 3',4 9'-tetrahydrospiro[indoline-3, 1 '- pyrido[3, 4-b]indol]-2-one is useful in the treatment and/or prevention of infections such as those caused by Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, Trypanosoma cruzi and parasites of the Leishmania genus such as, for example, Leishmania donovani., and it has the following structure:

(1 'R, 3'S)-5, 7'-dichloro-6'-fluoro-3'-methyl-2 3', 4', 9'-tetrahydrospiro[indoline-3, 1 - pyhdo[3, 4-b]indol]-2-one and a synthesis thereof are described in WO 2009/132921 Al in particular in Example 49 therein.

Example 10: Process for Conversion of Compound (IA) to Compound (IIA) in 30g Scale
![Figure imgf000101_0001]() 458.97
458.97
152.48g /so-propylamine hydrochloride and 0.204g pyridoxalphosphate monohydrate were dissolved in 495ml water while stirring. To this yellow clear solution a solution of 30. Og ketone in 85ml poly ethylene glycol (average mol weight 200) within 15 minutes. Upon addition the ketone precipitates as fine particles which are evenly distributed in the reaction media. To the suspension 180ml triethanolamine buffer (0.1 mol/l, pH 7) were added and the pH was adjusted to 7 by additon of aqueous sodium hydroxide solution (1 mol/l). The reaction mixture is heated to 50°C and a solution of 1.62g transaminase SEQ ID NO: 134 dissolved in 162ml triethanolamine buffer (0 1 mol/l, pH 7) is added. The reaction mixture is continiously kept at pH 7 by addition of 1 mol/l aqueous sodium hydroxide solution. The reaction mixture is stirred 24h at 50°C and a stream of Nitrogen is blown over the surface of the reaction mixture to strip off formed acetone. The reaction mixture is then cooled to 25°C and filtered over a bed of cellulose flock. The pH of the filtrate is adjusted to «1 by addition of concentrated sulfuric acid. The acidified filtrated is extracted with 250 ml /so-Propyl acetate. The layers are separated and the pH of the aqueous phase is adjusted to ¾10 by additon of concentrated aqueous sodium hydroxide solution. The basified aqueous phase is extracted with /'so-propyl acetate. The layers are seperated and the organic phase is washed with 100 ml water. The organic phase is concentrated by distillation to 2/3 of its origin volume. In a second reactor 33.98g (+)- camphor sulfonic acid is dissolved in 225 ml /'so-propyl acetate upon refluxing and the concentrated organic phase is added within 10 minutes. After complete addition the formed thin suspension is cooled to 0°C within 2 hours and kept at 0°C for 15 hours. The precipitated amine-(+)-camphor sulfonate salt is filtered, washed with 70 ml /so-propyl acetate and dried at 40°C in vaccuum yielding 51.57g of colourless crystals (84.5% yield t.q.)
Analytical Data
IR:
v (crn 1)=3296, 3061 , 2962, 2635, 2531 , 2078, 1741 , 1625, 1577, 1518, 1461 , 1415, 1392, 1375, 1324, 1302, 1280, 1256, 1226, 1 170, 1 126, 1096, 1041 , 988, 966, 937, 868, 834, 814, 790, 766, 746, 719, 669, 615.
LC-MS (ESI +):
Ammonium ion: m/z =227 ([M+H]), 268 ([M+H+CH3CN]), 453 ([2M+H]).
Camphorsulfonate ion: m/z =250 ([M+NH4]), 482 ([2M+NH4]).
LC-MS (ESI -):
Camphorsulfonate ion: m/z=231 ([M-H]), 463 ([2M-H]).
1H-NMR (DMSO-d6, 400 MHz):
1 1.22 (br. s., 1 H), 7.75 (br. s., 3H), 7.59 (d, J = 10.3 Hz, 1 H), 7.54 (d, J = 6.5 Hz, 1 H), 7.36 (d, J = 2.3 Hz, 1 H), 3.37 - 3.50 (m, 1 H), 2.98 (dd, J = 14.3, 5.8 Hz, 1 H), 2.91 (d, J = 14.8 Hz, 1 H), 2,81 (dd, J = 14.3, 8.0 Hz, 1 H), 2.63 - 2.74 (m, 1 H), 2.41 (d, J = 14.6 Hz, 1 H), 2.24 (dt, J = 18.3, 3.8 Hz, 1 H), 1 .94 (t, J = 4.4 Hz, 1 H), 1.86 (dt, J = 7.4, 3 6 Hz, 1 H), 1.80 (d, J = 18.1 Hz, H), 1.23 - 1 .35 (m, 2H), 1.15 (d, J = 6.3 Hz, 3H), 1.05 (s, 3H), 0.74 (s, 3H)
Free Amine (obtained by evaporatig the iso-Propylacetate layer after extraction of the basified aqueous layer):
1H NMR (400MHz, DMSO-d6): 11 .04 (br. s., 1 H), 7.50 (d, J = 10.5 Hz, 1 H), 7.48 (d, J = 6.5 Hz, 1 H), 7.25 (s, 1 H), 3.03 (sxt, J = 6.3 Hz, 1 H), 2.61 (dd, J - 14.3, 6.5 Hz, 1 H), 2.57 (dd, J = 14.1 , 6.5 Hz, 1 H), 1.36 (br. s., 2H), 0.96 (d, J = 6.3 Hz, 3H)
Example 11: Process for Conversion of Compound (HA) to Compound (IVB)
![Figure imgf000103_0001]() 3. solvent exchange to TP
3. solvent exchange to TP
13.62 g 5-chloroisatin is suspended in 35 ml /so-propanol and 2.3 g triethyl amine is added. The suspension is heated to reflux and a solution of 34.42g amine-(+)-camphor sulfonate salt dissolved in 300 ml /'so-propanol is added within 50 minutes. The reaction mixture is stirred at reflux for 17 hours. The reaction mixture is cooled to 75°C and 17.4g (+)-camphorsulfonic acid are added to the reaction mixture. Approximately 300 ml /so- propanol are removed by vacuum distillation. Distilled off /so-propanol is replaced by iso- propyl acetate and vacuum distillation is continued. This is distillation is repeated a second time. To the distillation residue 19 ml ethanol and 265 ml ethyl acetate is added and the mixture is heated to reflux. The mixture is cooled in ramps to 0°C and kept at 0°C for 24 hours. The beige to off white crystals are filtered off, washed with 3 portions (each 25 ml) precooled (0°C) ethylacetate and dried in vacuum yielding 40.3 g beige to off white crystals. (86.3% yield t.q.)
IR:
v (crrr)= 3229, 3115, 3078, 3052, 2971 , 2890, 2841. 2772. 2722, 2675, 2605, 2434. 1741 , 1718, 1621 , 1606, 1483, 1460, 1408, 1391 , 1372, 1336, 1307, 1277, 1267, 1238, 1202, 1 184, 1 162, 1 149, 1 128, 1067, 1036, 987, 973, 939, 919, 896, 871 , 857, 843, 785, 771 , 756, 717, 690, 678, 613.
LC-MS (ESI +):
Ammonium ion: m/z =390 ([M+H]), 431 ([M+H+CH3CN]) Camphorsulfonate ion: m/z =250 ([M+NH4]), 482 ([2M+NH4])
LC-MS (ESI -):
Camphorsulfonate ion: m/z=231 ([M-H]), 463 ([2M-H])
1H NMR (DMSO-d6, 600 MHz):
11.49 (s, 1 H), 1 1.23 (s, 1 H), 10.29 - 10.83 (m, 1 H), 9.78 - 10.31 (m, 1 H), 7.55 - 7.60 (m, 2H), 7.52 (s, 1 H), 7.40 (d, J = 6.2 Hz, H), 7.16 (d, J = 8.8 Hz, 1 H), 4.52 - 4.63 (m, 1 H). 3.20 (dd, J = 16.3, 4.2 Hz, 1 H), 2.96 (dd, J = 16.1 , 11.3 Hz, 1 H), 2.90 (d, J = 15.0 Hz, 1 H), 2.56 - 2.63 (m, 1 H), 2.39 (d, J = 14.6 Hz, 1 H), 2.21 (dt, J = 18.0, 3.8 Hz, 1 H), 1.89 - 1.93 (m, 1 H), 1.81 (ddd, J = 15.3, 7.8, 3.7 Hz, 1 H), 1.76 (d, J = 18.3 Hz, 1 H), 1 .53 (d, J = 6.6 Hz, 3H), 1.20 - 1.33 (m, 2H), 0.98 (s, 3H), 0.70 (s, 3H)
Example 12: Process for Preparing a Compound of formula (IVA) 1/z Hydrate
![Figure imgf000104_0001]() mw622.54 .............................................................................mw399.25
mw622.54 .............................................................................mw399.25
In a 750ml reactor with impeller stirrer 50g of compound (IVB) salt were dissolved in 300ml Ethanol (ALABD) and 100 ml deionised Water (WEM). The clear, yellowish sollution was heated to 58°C internal temperature. To the solution 85 g of a 10% aqueous sodium carbonate solution was added within 10 minutes. The clear solution was particle filtered into a second reaction vessel. Vessel and particle filter were each rinsed with 25 ml of a mixture of ethanohwater (3:1 v/v) in the second reaction vessel. The combined particle filtered solution is heated to 58°C internal temperature and 200ml water (WEM) were added dropwise within 15 minutes. Towards the end of the addition the solution gets turbid.
The mixture is stirred for 10 minutes at 58°C internal temperature and is then cooled slowely to room temperature within 4hours 30 minutes forming a thick, well stirable white suspension. To the suspension 200 ml water are added and the mixture is stirred for additional 15hours 20 minutes at room temperature. The suspension is filtered and the filter cake is washed twice with 25 ml portions of a mixture of ethanohwater 9: 1 (v/v). The colourless crystals are dried at 60°C in vacuum yielding 26.23g (=91.2% yield). H NMR (400 MHz, DMSO-d6)
0.70 (s, 1H), 10.52 (s, 1H), 7.44 (d, J = 10.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H),.26 (d, J = 6.5 Hz, 1H), 7.05 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 3.83 - 4.00 (m,H), 3.13 (d, J = 6.0 Hz, 1H), 2.77 (dd, J = 15.1, 3.8 Hz, 1H), 2.38 (dd, J = 15.1, 10.5 Hz,H), 1.17 (d, J = 6.3 Hz, 3H).

152.48g /so-propylamine hydrochloride and 0.204g pyridoxalphosphate monohydrate were dissolved in 495ml water while stirring. To this yellow clear solution a solution of 30. Og ketone in 85ml poly ethylene glycol (average mol weight 200) within 15 minutes. Upon addition the ketone precipitates as fine particles which are evenly distributed in the reaction media. To the suspension 180ml triethanolamine buffer (0.1 mol/l, pH 7) were added and the pH was adjusted to 7 by additon of aqueous sodium hydroxide solution (1 mol/l). The reaction mixture is heated to 50°C and a solution of 1.62g transaminase SEQ ID NO: 134 dissolved in 162ml triethanolamine buffer (0 1 mol/l, pH 7) is added. The reaction mixture is continiously kept at pH 7 by addition of 1 mol/l aqueous sodium hydroxide solution. The reaction mixture is stirred 24h at 50°C and a stream of Nitrogen is blown over the surface of the reaction mixture to strip off formed acetone. The reaction mixture is then cooled to 25°C and filtered over a bed of cellulose flock. The pH of the filtrate is adjusted to «1 by addition of concentrated sulfuric acid. The acidified filtrated is extracted with 250 ml /so-Propyl acetate. The layers are separated and the pH of the aqueous phase is adjusted to ¾10 by additon of concentrated aqueous sodium hydroxide solution. The basified aqueous phase is extracted with /'so-propyl acetate. The layers are seperated and the organic phase is washed with 100 ml water. The organic phase is concentrated by distillation to 2/3 of its origin volume. In a second reactor 33.98g (+)- camphor sulfonic acid is dissolved in 225 ml /'so-propyl acetate upon refluxing and the concentrated organic phase is added within 10 minutes. After complete addition the formed thin suspension is cooled to 0°C within 2 hours and kept at 0°C for 15 hours. The precipitated amine-(+)-camphor sulfonate salt is filtered, washed with 70 ml /so-propyl acetate and dried at 40°C in vaccuum yielding 51.57g of colourless crystals (84.5% yield t.q.)
Analytical Data
IR:
v (crn 1)=3296, 3061 , 2962, 2635, 2531 , 2078, 1741 , 1625, 1577, 1518, 1461 , 1415, 1392, 1375, 1324, 1302, 1280, 1256, 1226, 1 170, 1 126, 1096, 1041 , 988, 966, 937, 868, 834, 814, 790, 766, 746, 719, 669, 615.
LC-MS (ESI +):
Ammonium ion: m/z =227 ([M+H]), 268 ([M+H+CH3CN]), 453 ([2M+H]).
Camphorsulfonate ion: m/z =250 ([M+NH4]), 482 ([2M+NH4]).
LC-MS (ESI -):
Camphorsulfonate ion: m/z=231 ([M-H]), 463 ([2M-H]).
1H-NMR (DMSO-d6, 400 MHz):
1 1.22 (br. s., 1 H), 7.75 (br. s., 3H), 7.59 (d, J = 10.3 Hz, 1 H), 7.54 (d, J = 6.5 Hz, 1 H), 7.36 (d, J = 2.3 Hz, 1 H), 3.37 - 3.50 (m, 1 H), 2.98 (dd, J = 14.3, 5.8 Hz, 1 H), 2.91 (d, J = 14.8 Hz, 1 H), 2,81 (dd, J = 14.3, 8.0 Hz, 1 H), 2.63 - 2.74 (m, 1 H), 2.41 (d, J = 14.6 Hz, 1 H), 2.24 (dt, J = 18.3, 3.8 Hz, 1 H), 1 .94 (t, J = 4.4 Hz, 1 H), 1.86 (dt, J = 7.4, 3 6 Hz, 1 H), 1.80 (d, J = 18.1 Hz, H), 1.23 - 1 .35 (m, 2H), 1.15 (d, J = 6.3 Hz, 3H), 1.05 (s, 3H), 0.74 (s, 3H)
Free Amine (obtained by evaporatig the iso-Propylacetate layer after extraction of the basified aqueous layer):
1H NMR (400MHz, DMSO-d6): 11 .04 (br. s., 1 H), 7.50 (d, J = 10.5 Hz, 1 H), 7.48 (d, J = 6.5 Hz, 1 H), 7.25 (s, 1 H), 3.03 (sxt, J = 6.3 Hz, 1 H), 2.61 (dd, J - 14.3, 6.5 Hz, 1 H), 2.57 (dd, J = 14.1 , 6.5 Hz, 1 H), 1.36 (br. s., 2H), 0.96 (d, J = 6.3 Hz, 3H)
Example 11: Process for Conversion of Compound (HA) to Compound (IVB)

13.62 g 5-chloroisatin is suspended in 35 ml /so-propanol and 2.3 g triethyl amine is added. The suspension is heated to reflux and a solution of 34.42g amine-(+)-camphor sulfonate salt dissolved in 300 ml /'so-propanol is added within 50 minutes. The reaction mixture is stirred at reflux for 17 hours. The reaction mixture is cooled to 75°C and 17.4g (+)-camphorsulfonic acid are added to the reaction mixture. Approximately 300 ml /so- propanol are removed by vacuum distillation. Distilled off /so-propanol is replaced by iso- propyl acetate and vacuum distillation is continued. This is distillation is repeated a second time. To the distillation residue 19 ml ethanol and 265 ml ethyl acetate is added and the mixture is heated to reflux. The mixture is cooled in ramps to 0°C and kept at 0°C for 24 hours. The beige to off white crystals are filtered off, washed with 3 portions (each 25 ml) precooled (0°C) ethylacetate and dried in vacuum yielding 40.3 g beige to off white crystals. (86.3% yield t.q.)
IR:
v (crrr)= 3229, 3115, 3078, 3052, 2971 , 2890, 2841. 2772. 2722, 2675, 2605, 2434. 1741 , 1718, 1621 , 1606, 1483, 1460, 1408, 1391 , 1372, 1336, 1307, 1277, 1267, 1238, 1202, 1 184, 1 162, 1 149, 1 128, 1067, 1036, 987, 973, 939, 919, 896, 871 , 857, 843, 785, 771 , 756, 717, 690, 678, 613.
LC-MS (ESI +):
Ammonium ion: m/z =390 ([M+H]), 431 ([M+H+CH3CN]) Camphorsulfonate ion: m/z =250 ([M+NH4]), 482 ([2M+NH4])
LC-MS (ESI -):
Camphorsulfonate ion: m/z=231 ([M-H]), 463 ([2M-H])
1H NMR (DMSO-d6, 600 MHz):
11.49 (s, 1 H), 1 1.23 (s, 1 H), 10.29 - 10.83 (m, 1 H), 9.78 - 10.31 (m, 1 H), 7.55 - 7.60 (m, 2H), 7.52 (s, 1 H), 7.40 (d, J = 6.2 Hz, H), 7.16 (d, J = 8.8 Hz, 1 H), 4.52 - 4.63 (m, 1 H). 3.20 (dd, J = 16.3, 4.2 Hz, 1 H), 2.96 (dd, J = 16.1 , 11.3 Hz, 1 H), 2.90 (d, J = 15.0 Hz, 1 H), 2.56 - 2.63 (m, 1 H), 2.39 (d, J = 14.6 Hz, 1 H), 2.21 (dt, J = 18.0, 3.8 Hz, 1 H), 1.89 - 1.93 (m, 1 H), 1.81 (ddd, J = 15.3, 7.8, 3.7 Hz, 1 H), 1.76 (d, J = 18.3 Hz, 1 H), 1 .53 (d, J = 6.6 Hz, 3H), 1.20 - 1.33 (m, 2H), 0.98 (s, 3H), 0.70 (s, 3H)
Example 12: Process for Preparing a Compound of formula (IVA) 1/z Hydrate

In a 750ml reactor with impeller stirrer 50g of compound (IVB) salt were dissolved in 300ml Ethanol (ALABD) and 100 ml deionised Water (WEM). The clear, yellowish sollution was heated to 58°C internal temperature. To the solution 85 g of a 10% aqueous sodium carbonate solution was added within 10 minutes. The clear solution was particle filtered into a second reaction vessel. Vessel and particle filter were each rinsed with 25 ml of a mixture of ethanohwater (3:1 v/v) in the second reaction vessel. The combined particle filtered solution is heated to 58°C internal temperature and 200ml water (WEM) were added dropwise within 15 minutes. Towards the end of the addition the solution gets turbid.
The mixture is stirred for 10 minutes at 58°C internal temperature and is then cooled slowely to room temperature within 4hours 30 minutes forming a thick, well stirable white suspension. To the suspension 200 ml water are added and the mixture is stirred for additional 15hours 20 minutes at room temperature. The suspension is filtered and the filter cake is washed twice with 25 ml portions of a mixture of ethanohwater 9: 1 (v/v). The colourless crystals are dried at 60°C in vacuum yielding 26.23g (=91.2% yield). H NMR (400 MHz, DMSO-d6)
0.70 (s, 1H), 10.52 (s, 1H), 7.44 (d, J = 10.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.1 Hz, 1H),.26 (d, J = 6.5 Hz, 1H), 7.05 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.3 Hz, 1H), 3.83 - 4.00 (m,H), 3.13 (d, J = 6.0 Hz, 1H), 2.77 (dd, J = 15.1, 3.8 Hz, 1H), 2.38 (dd, J = 15.1, 10.5 Hz,H), 1.17 (d, J = 6.3 Hz, 3H).
......................................................
Journal of Medicinal Chemistry, 2010 , vol. 53, 14 p. 5155 - 5164
(1R,3S)-5′,7-Dichloro-6-fluoro-3-methyl-2,3,4,9-tetrahydrospiro[β-carboline-1,3′-indol]-2′(1′H)-one (19a)
1H NMR (500 MHz, DMSO-d6): δ 10.69 (s, 1H), 10.51 (s, 1H), 7.43 (d, J = 10.0 Hz, 1H), 7.33 (dd, J = 8.0, 2.2 Hz, 1H), 7.27 (d, J = 6.5 Hz, 1H), 7.05 (d, J = 2.3 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 3.91 (m, 1H), 3.13 (bd, J = 6.2 Hz, 1H), 2.74 (dd, J = 15.0, 3.0 Hz, 1H), 2.35 (dd, J = 15.0, 10.3 Hz, 1H), 1.15 (d, J = 6.0 Hz, 3H). MS (ESI) m/z 392.0 (M + 2H)+; [α]D25 = +255.4° (c = 0.102 g/L, methanol).
......................................
Z.Zhang, WO 2007 / 104714,2007).
[0007]
![Figure CN102432526AD00051]() [0008] (2) year 2008 Roche pharmaceutical company disclosed a spiro [oxindole - cyclohexenone] skeleton biomedicine, PCT International Application No. W02008 / 055812. It also announced the preparation of anti-cancer agents and antagonists of the application of the compound is used as the interaction with MDM2 (reference:. Liu, J.-J; Zhang, Z; (Hoffmann-LaRoche AG), PCT Int App 1. . W02008 / 055812, 2008), its structural formula is as follows:
[0008] (2) year 2008 Roche pharmaceutical company disclosed a spiro [oxindole - cyclohexenone] skeleton biomedicine, PCT International Application No. W02008 / 055812. It also announced the preparation of anti-cancer agents and antagonists of the application of the compound is used as the interaction with MDM2 (reference:. Liu, J.-J; Zhang, Z; (Hoffmann-LaRoche AG), PCT Int App 1. . W02008 / 055812, 2008), its structural formula is as follows:
[0009]
![Figure CN102432526AD00052]() [0010] (3) Melchiorre research group abroad chiral amines and o-fluoro-3-benzyl benzoate as catalyst methylene-indole-2-one (3-benzylideneindolin-2-one, CAS Number: 3359-49- 7) with α, β - unsaturated ketone synthesis of chiral spiro [cyclohexane _1,3'- indole] _2,4 '- dione [s pir0 [cycl0hexane-l, 3' -indoline] - 2 ', 4-diones] compounds (see:.. Bencivenni, G; ffu, LY; Mazzanti, A .; Giannichi, B.; Pesciaioli, F; Song, Μ P.; Bartoli, G.; Melchiorre, P .... .Angew Chem Int Ed 2009,48,7200), the structure of the total formula is as follows:
[0010] (3) Melchiorre research group abroad chiral amines and o-fluoro-3-benzyl benzoate as catalyst methylene-indole-2-one (3-benzylideneindolin-2-one, CAS Number: 3359-49- 7) with α, β - unsaturated ketone synthesis of chiral spiro [cyclohexane _1,3'- indole] _2,4 '- dione [s pir0 [cycl0hexane-l, 3' -indoline] - 2 ', 4-diones] compounds (see:.. Bencivenni, G; ffu, LY; Mazzanti, A .; Giannichi, B.; Pesciaioli, F; Song, Μ P.; Bartoli, G.; Melchiorre, P .... .Angew Chem Int Ed 2009,48,7200), the structure of the total formula is as follows:
[0011]
![Figure CN102432526AD00061]() [0012] (4) Gong Flow column team found to cyclohexanediamine derived Bronsted acid - a bifunctional catalyst Lewis base catalysis of 3-benzyl-methylene-indole-2-one and α, β- unsaturated 1,3 tandem reaction dicarbonyl compound (Nazarov reagent) can be obtained with high stereoselectivity chiral spiro [cyclohexane _1,3'- indol] -2 ', 4-dione [spiro [cyclohexane-l, 3 '-indoline] -2', 4-diones] compounds; and by this method successfully synthesized 7 Roche pharmaceutical companies to develop chiral anti-tumor agents (see: Q Wei, L -Z Gong, Org Lett 2010..... , 12, 1008.).
[0012] (4) Gong Flow column team found to cyclohexanediamine derived Bronsted acid - a bifunctional catalyst Lewis base catalysis of 3-benzyl-methylene-indole-2-one and α, β- unsaturated 1,3 tandem reaction dicarbonyl compound (Nazarov reagent) can be obtained with high stereoselectivity chiral spiro [cyclohexane _1,3'- indol] -2 ', 4-dione [spiro [cyclohexane-l, 3 '-indoline] -2', 4-diones] compounds; and by this method successfully synthesized 7 Roche pharmaceutical companies to develop chiral anti-tumor agents (see: Q Wei, L -Z Gong, Org Lett 2010..... , 12, 1008.).
[0013] (5) Wang Lixin research group recently reported that primary amines derived from cinchona alkaloids and Bronsted acid as catalyst N- protected indolone compounds and double Michael addition reaction of diketene generate hand spiro [cyclohexane-1, 3'-indol] -2 ', 4-dione [spiro [cyclohexane-l, 3' -indoline] -2 ', 4-diones] type of tx ^ (: L. -L. Wang, L. Peng, J. -F. Bai, L. -N. Jia, X. -Y. Luo, QC Huang, L. -X. Wang, Chem. Commum. 2011,47, 5593.).
[0007]

[0009]

[0011]

[0013] (5) Wang Lixin research group recently reported that primary amines derived from cinchona alkaloids and Bronsted acid as catalyst N- protected indolone compounds and double Michael addition reaction of diketene generate hand spiro [cyclohexane-1, 3'-indol] -2 ', 4-dione [spiro [cyclohexane-l, 3' -indoline] -2 ', 4-diones] type of tx ^ (: L. -L. Wang, L. Peng, J. -F. Bai, L. -N. Jia, X. -Y. Luo, QC Huang, L. -X. Wang, Chem. Commum. 2011,47, 5593.).
References![]()
![]()
![]()
![]()
- "NITD 609". Medicines for Malaria Venture.
- Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, González-Páez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT (2010). "Spiroindolones, a potent compound class for the treatment of malaria". Science329 (5996): 1175–80. doi:10.1126/science.1193225. PMC 3050001. PMID 20813948.
| WO2009132921A1 * | Apr 1, 2009 | Nov 5, 2009 | Novartis Ag | Spiro-indole derivatives for the treatment of parasitic diseases |
| WO2010081053A2 * | Jan 8, 2010 | Jul 15, 2010 | Codexis, Inc. | Transaminase polypeptides |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| AT507050A1 * | Title not available | |||
| EP0036741A2 * | Mar 17, 1981 | Sep 30, 1981 | THE PROCTER & GAMBLE COMPANY | Phosphine compounds, transition metal complexes thereof and use thereof as chiral hydrogenation catalysts |
| EP0120208A2 * | Jan 24, 1984 | Oct 3, 1984 | Degussa Aktiengesellschaft | Microbiologically produced L-phenylalanin-dehydrogenase, process for obtaining it and its use |
| EP0135846A2 * | Aug 31, 1984 | Apr 3, 1985 | Genetics Institute, Inc. | Production of L-amino acids by transamination |
| GB974895A * | Title not available | |||
| US3282959 * | Mar 21, 1962 | Nov 1, 1966 | Parke Davis & Co | 7-chloro-alpha-methyltryptamine derivatives |
| US4073795 * | Jun 22, 1976 | Feb 14, 1978 | Hoffmann-La Roche Inc. | Synthesis of tryptophans |
| WO2005009370A2 * | Jul 22, 2004 | Feb 3, 2005 | Pharmacia Corp | Beta-carboline compounds and analogues thereof and their use as mitogen-activated protein kinase-activated protein kinase-2 inhibitors |
| EP0466548A1 * | Jun 27, 1991 | Jan 15, 1992 | Adir Et Compagnie | 1,2,3,4,5,6-Hexahydroazepino[4,5-b]indole and 1,2,3,4-tetrahydro-beta-carbolines, processes for their preparation, and pharmaceutical compositions containing them |
Рисунок из Science 2010, 329, 1175
Исследовательская группа Элизабет Винцелер (Elizabeth A. Winzeler) разработала новый препарат, первоначально проведя скрининг библиотеки, состоящей из 12000 соединений, а затем получив производные наиболее перспективных кандидатов. В результате долгой работы исследователи отобрали единственное соединение спироиндолоновой структуры, получившее регистрационный номер NITD609. В случае успешного прохождения экспертизы фармакологических и токсикологических свойств нового соединения исследователи надеются приступить к первой фазе его клинических испытаний уже в конце этого года.
Было обнаружено, что NITD609быстро останавливает белковый синтез в организме возбудителя малярии, ингибируя ген аденозинтрифосфатазы, ответственной за транспорт катионов через мембрану клетки возбудителя. То, что механизм действия нового соединения отличается от механизма, характерного для других средств лечения малярии, объясняет причины успешного действия нового препарата в том числе и против штаммов малярии, выработавших резистентность.
HPLC
Analyte quantization was performed byLC/MS/MS. Liquid chromatography was performed using an Agilent
1100 HPLC system(Santa Clara, CA), with the Agilent Zorbax XDB Phenyl (3.5μ, 4.6 x75 mm) column at
an oven temperature of 35 °C, coupled with a QTRAP4000 triple quadruple mass
spectrometer (Applied Biosystems, Foster City, CA). Instrumentcontrol and dataacquisition were performed using Applied Biosystems software Analyst 1.4.2. Themobile phases used were A: water:acetic acid (99.8:0.2, v/v) and B: acetonitrile:aceticacid (99.8:0.2, v/v), using a gradient, with flow rate of 1.0 mL/min, and run time of 5minutes. Under these conditions the retention time of9a
was 3.2 minutes. Compounddetection on the mass spectrometer was performed in electrospraypositive ionizationmode and utilized multiple reaction monitoring (MRM) for specificity (9atransitions338.3/295.1, 338.3/259.2) together with their optimized MS parameters. The lower limitof quantification for9awas 70 ng/mL.
Extraction and LCMS analysis of 20a.Plasma samples were extracted withacetonitrile:methanol-acetic acid (90:9.8:0.2 v/v) for the analyte and internal standard(17a) using a 3.6 to 1 extractant to plasma ratio. Analyte quantitation was performed by
LC/MS/MS. Liquid chromatography was performed using an Agilent1100 HPLC systemS7(Santa Clara, CA), with the Agilent Zorbax XDB-Phenyl (3.5μ, 4.6x75mm) column atan oven temperature of 45 °C coupled with a QTRAP 4000 triple quadruple massSpectrometer (Applied Biosystems, Foster City, CA). Instrumentcontrol and dataacquisition were performed using Applied Biosystems software Analyst 1.4.2. Themobile phases used were A: water:acetic acid (99.8:0.2, v/v) and B: methanol:acetic acid
(99.8:0.2, v/v), using gradient elution conditions with a flow rate of 1.0 mL/min and a runtime of 6 minutes
↧
↧
Dorzolamide Hydrochloride

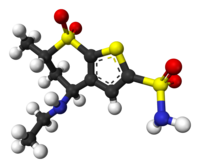
Trusopt, 120279-96-1, 1cil, Trusopt (TN), Dorzolamide (DZA), Dorzolamide (INN), MK507
Molecular Formula:C10H16N2O4S3
Molecular Weight:324.44004 g/mol
(4S,6S)-4-(ethylamino)-6-methyl-7,7-dioxo-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide
(4S,6S)-4-(ethylamino)-5,6-dihydro-6-methyl-4H- thieno[2,3-/?]thiopyran-2-sulfonamide 7,7-dioxide
(4S-trans)-4-(ETHYLAMINO)-5,6-dihydro-6-methyl-4H-thieno(2,3-b)thiopyran-2-sulfonamide-7,7-dioxide
Antiglaucoma Agents, OCULAR MEDICATIONS, Ophthalmic Drugs, Carbonic Anhydrase Inhibitors
| HS CODE: | 2935009090 |
|---|
120279-96-1
130693-82-2..HCL
Laszlo Kovacs, Csaba Szabo, Erika Molnarne, Adrienne Kovacsne-Mezei, Claude Singer, Judith Aronhime, “Method of making dorzolamide hydrochloride.” U.S. Patent US20060155132, issued July 13, 2006.
Dorzolamide is a carbonic anhydrase (CA) inhibitor. It is used in ophthalmic solutions (Trusopt) to lower intraocular pressure (IOP) in open-angle glaucoma and ocular hypertension.
Dorzolamide (trade name Trusopt) is a carbonic anhydrase inhibitor. It is ananti-glaucoma agent, and acts by decreasing the production of aqueous humour.[1] It is optically applied in the form of a 2% eye drops.[2]
History
This drug, developed by Merck, was the first drug in human therapy (market introduction 1995) which resulted from structure-baseddrug design. It was developed to circumvent the systemic side effects of acetazolamide which has to be taken orally.[2]
Uses
Dorzolamide hydrochloride is used to lower increased intraocular pressure in open-angle glaucoma and ocular hypertension.
Pharmacodynamics
Side effects
Ocular stinging, burning, itching and bitter taste.[2] it causes shallowing of the anterior chamber and leads to transient Myopia.
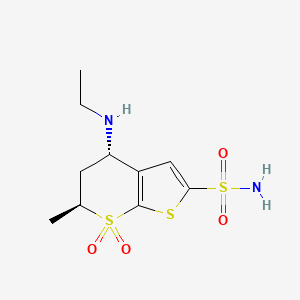
Title: Dorzolamide
CAS Registry Number: 120279-96-1
CAS Name: (4S,6S)-4-(Ethylamino)-5,6-dihydro-6-methyl-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide
Molecular Formula: C10H16N2O4S3
Molecular Weight: 324.44
Percent Composition: C 37.02%, H 4.97%, N 8.63%, O 19.73%, S 29.65%
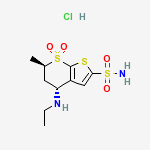
Derivative Type: Hydrochloride
CAS Registry Number: 130693-82-2
Manufacturers’ Codes: MK-507
Trademarks: Trusopt (Merck & Co.)
Molecular Formula: C10H16N2O4S3.HCl
Molecular Weight: 360.90
Percent Composition: C 33.28%, H 4.75%, N 7.76%, O 17.73%, S 26.65%, Cl 9.82%
Properties: mp 283-285°. [a]D24 -8.34° (c = 1 in methanol). Sol in water.
Melting point: mp 283-285°
Optical Rotation: [a]D24 -8.34° (c = 1 in methanol)
Dorzolamide Hydrochloride and its derivatives is known. U.S. Pat. No. 5,688,968 describes preparation of Dorzolamide HCl starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7-dioxide, as depicted in scheme 1:

The process described in BP 0 296 879 (equivalent of U.S. Pat. No. 4,797,413) is of particular relevance. EP 0 296 879 describes the synthesis of Dorzolamide Hydrochloride starting from thiophene-2-thiol as depicted in scheme 2 and 3



The process described in EP 0,296,879 (scheme 2) has the following disadvantages: (a) The starting material Thiophene-2-thiol is unstable and undergoes oxidation to form disulfide, leading to lower yield of viii; (b) the yield of sulfonamide (xii) from sulphonic acid (x) is very poor (35%) and requires use of 18-crown-6 ether, which is expensive; (c) oxidation of alcohol (xiii) to sulfone is carried out using oxone which is expensive and hazardous; and separation of cis/trans isomer is done by column chromatography which is industrially inconvenient.
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (4S,6S)-2-ethylamino-4-methyl-5,5-dioxo- 5λ6,7-dithiabicyclo[4.3.0]nona-8,10-diene-8-sulfonamide | |
| CLINICAL DATA | |
| TRADE NAMES | Trusopt |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a602022 |
| PREGNANCY CATEGORY |
|
| LEGAL STATUS | |
| ROUTES | Topical (eye drops) |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | ~33% |
| HALF-LIFE | 4 months |
| IDENTIFIERS | |
| CAS NUMBER | 130693-82-2 120279-96-1 |
| ATC CODE | S01EC03 |
| PUBCHEM | CID 5284549 |
| DRUGBANK | DB00869 |
| CHEMSPIDER | 4447604 |
| UNII | 9JDX055TW1 |
| KEGG | D07871 |
| CHEBI | CHEBI:4702 |
| CHEMBL | CHEMBL218490 |
| CHEMICAL DATA | |
| FORMULA | C10H16N2O4S3 |
| MOLECULAR MASS | 324.443 g/mol |
TRUSOPT® (dorzolamide hydrochloride ophthalmic solution) is a carbonic anhydrase inhibitor formulated for topical ophthalmic use.
Dorzolamide hydrochloride is described chemically as: (4S-trans)-4-(ethylamino)-5,6-dihydro-6methyl-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide monohydrochloride. Dorzolamide hydrochloride is optically active. The specific rotation is
 |
Its empirical formula is C10H16N2O4S3•HCl and its structural formula is:
 |
Dorzolamide hydrochloride has a molecular weight of 360.9 and a melting point of about 264°C. It is a white to off-white, crystalline powder, which is soluble in water and slightly soluble in methanol and ethanol.
TRUSOPT Sterile Ophthalmic Solution is supplied as a sterile, isotonic, buffered, slightly viscous, aqueous solution of dorzolamide hydrochloride. The pH of the solution is approximately 5.6, and the osmolarity is 260-330 mOsM. Each mL of TRUSOPT 2% contains 20 mg dorzolamide (22.3 mg of dorzolamide hydrochloride). Inactive ingredients are hydroxyethyl cellulose, mannitol, sodium citrate dihydrate, sodium hydroxide (to adjust pH) and water for injection. Benzalkonium chloride 0.0075% is added as a preservative.
…………………………………………
The dorzolamide hydrochloride product is prepared from the aminated intermediate of Formula IV by the following scheme.


[00056] Preparation of dorzolamide hydrochloride product from the animated intermediate of Formula IV
[00057] Fuming sulfuric acid (20%, 5 1) is cooled to -7°±2°C and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°+5°C during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5°C. Thionyl chloride (20 1) is added to the stirred reaction mixture at 20±5°C. The reaction mixture is heated to 60°-65°C and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2°C and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ~9 1.) The residue is cooled to -5°+2°C.
[00058] Ethyl acetate (75 1) is cooled to -10°±5°C and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25°C. Aqueous ammonia (25%, 75 1) is cooled to -10°±5°C and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 300C. The final pH: ~11. The slurry is cooled to 0°+2°C and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2x 20 1 and 10 1). Ethyl acetate is evaporated from the filtrate at 38°±2°C under vacuum. The residue is heated to 38°±2°C, washed with toluene (3×37.5 1) at this temperature. Water (25 1) is added to the aqueous phase, cooled to 20°-25°C and extracted with ethyl acetate (3x 75 1, 37.5 1, and 37.5 1). The collected ethyl acetate phase is concentrated to ~ 100 1 at 38°±2°C under vacuum. The residue is cooled to 20°-25°C and hydrogen chloride in ethanol (5%, 10.8 1) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 1) and dried at 55°-60°C under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (~2 Kg).
[00059] Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 1) at 20°-25°C and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 1). The formed slurry is extracted with ethyl acetate (5×72 1). The collected ethyl acetate phase is concentrated to 180 1 by vacuum distillation. The residue is cooled to 20°-25°C, ethyl acetate (45 1) and hydrogen chloride in ethanol (5%, 22.5 1) are added to it during stirring (pH:~1.0). The formed slurry is stirred for 1 hour at 20°-25°C then cooled to 0°-4°C and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 1), and dried at 55°-60°C under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (~8.2Kg).
[00060] Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water
(24 1) at 95°-105°C and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4°C and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 1) and dried at 55°-60°C under vacuum for 4-8 hours to give crystallized DRZ HCl salt (~6.6 Kg).
………………………………………………..
The invention provides a process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S)methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide hydrochloride of formula (I), comprising of nine steps, as depicted in scheme 4 below:


Example 8Preparation of Trans 5,6 dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2-sulfonamide-7,7 dioxide (X)A solution of product from example 7 (39.5 gm, 0.132 mole) in ethyl acetate (426 ml) was cooled to 0 to 5° C. and ethanolic HCl (20 ml) was added and stirred for 3 hrs at 0 to 5° C. The product was precipitated out, filtered and washed with chilled ethyl acetate. The cake was sucked to remove as much ethyl acetate as possible, and dried to get compound (21 gm) The product was suspended into ethyl acetate (210 ml), refluxed for 1 hr, then cooled to 10° C. The product was filtered and washed with chilled ethyl acetate. The cake was sucked to remove as much ethyl acetate as possible, and dried to hydrochloride salt of title compound (18 gm). The salt was then treated with saturated solution of sodium bicarbonate and mixture extracted with ethyl acetate. The organic extract were dried, filtered and concentrated to dryness to yield title compound (X) (15 gm, 37.98%).
Example 9Preparation of 5,6 dihydro-4H-4-(S)-ethylamino-6-(S)-methylthieno[2,3-b]thiopyran-2-sulfonamide-7,7 dioxide Hydrochloride (I)
A mixture of compound from example 8 (15 gm0.0462 mole) and di-p-toluyl-D-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) was heated to boiling and hot solution filtered through a filter-aid pad with a layer of charcoal. The filtrate was concentrated by boiling to a volume of about (400 ml) and then allowed to crystallize. After standing overnight the crystals were filtered off and material recrystallized twice more from n-propanol (400 ml) to yield a 2:1 salt of free base to acid. Combined mother liquors from this recrystallization were saved for stage B. The salt was then treated with a saturated solution of sodium bicarbonate and mid extracted with ethyl acetate. The organic extract were dried, filtered and concentrated to dryness to yield (3.2 gm) of freebase. The hydrochloride salt was prepared from 5,6 N HCl ethanol and crystallized from methanol-isopropanol to yield (2.83 gm) of (+) isomer, SOR 8.23 (C 0.9 methanol) M.P. 283-285° C. The combine mother liquor was treated with saturated solution of sodium bicarbonate and mixture extracted with ethyl acetate. The organic exacts were dried, filtered and concentrated to dryness. The residue was treated with di-p-toluyl-L-tartaric acid monohydrate (4.55 gm, 0.01125 mole) in n-propanol (1600 ml) and the isomer separated by the process described previously to give title compound (I) (3.75 gm, 22.48%) SOR=−8.34 (C 1, Methaol) M.P. 283 to 285° C.,
………………………………………..
Dorzolamide is chemically termed as (4S,6S)-4-(ethylamino)-5,6-dihydro-6-methyl-4H- thieno[2,3-/?]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride. Dorzolamide hydrochloride is represented by following structural Formula I:
HN ‘CH,

Formula I
Dorzolamide hydrochloride is known to be a carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
A process for the preparation of dorzolamide and its derivatives was first described in EP 0296879. The process of particular relevance is depicted in scheme 1. Scheme 1

(viϋ) (ix) Trans and Cis (x)

Trans (xi) Trans(+) (xii) ( I )
The process disclosed in scheme 1 has following disadvantages.
(a) The reduction of the ketone of sulfonamide (vi) using absolute ethanol is carried out at reflux and then stirred at room temperature for several hours to complete the reaction. This longer duration of reaction produces many impurities.
(b) Oxidation of alcohol (vii) to sulfone (viii) is carried out using oxone. The oxone has many disadvantages such as it is irritating to the eyes, skin, nose and throat. It should be used with adequate ventilation and exposure to its dust should be minimized. Traces of heavy metal salts catalyze the decomposition of oxone. It is practically insoluble in all organic solvents hence a phase transfer catalyst is required.
(c) Activation of the 4-hydroxy group of the sulfoaminated hydroxysulfone (viii) and nucleophilic substitution by desired ethylamine, results in all diastereomeric products (x) i.e. trans and cis isomers, which must be separated by column chromatography and resolved, further using resolving agent. As a result, product loss is greater when the desired product is the more active enantiomer.
An alternate route for the preparation of dorzolamide hydrochloride by the Ritter reaction is disclosed in EP0296879 and consists of the treatment of a aliphatic hydroxyl with a nitrile and a strong acid to form an amide. The process disclosed is as depicted in Scheme 2.
Scheme 2

(viii) (ix-a ) Trans and Cis (x)

Trans(+) (xii)
Trans (+/-) (xi) ( I )
The reaction involves conversion of hydroxysulfones (viii) to the corresponding acetoamidosulfones (ix-a) with retention of configuration followed by reduction of the amido group, chromatographic separation and resolution to obtain the desired trans isomer (I).
The prior art teaches the use of an excess quantity of sulfuric acid to carry out the Ritter reaction and hence a large quantity of ice is required for quenching the reaction mass. When the reaction mass in concentrated sulfuric acid comes into contact with ice, a large amount of localized heat is generated causing decomposition of material. Since a huge amount of water is required for quenching the reaction mass, the amount of ethyl acetate required for extraction is also substantially large. The work-up using water is not advisable nor applicable industrially.
United States Patent 5688968 describes an alternative route of preparation of dorzolamide hydrochloride starting from chiral 5,6-dihydro-4-(S)-hydroxy-6-(S)-methyl-4H-thiopyran-7,7- dioxide, as depicted in Scheme 3:
Scheme 3

(xiv) (XV)
(xiii)

(xvi) (xvii ) Trans:Cis:: 95: 5 (xviii)
HN CH,


(xix) ( I )
The process described in Scheme 3 has the following disadvantages: (a) Use of expensive chiral hydroxysulfone starting material. The process for the preparation of the chiral hydroxysulfone starting material is disclosed in U.S. Patents Nos. 5,157,129, 5,474,919 and 5,760,249. In these processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone, (b) The process according to this patent uses maleic acid to separate the undesired cis- isomer from dorzolamide. However this maleate salt formation to remove the cis isomer is only suitable when the ratio of trans/cis is greater than 95:5. That means, the maleate salt formation of dorzolamide does not the remove cis isomer exclusively when the cis isomer content is more than 5%. It sometimes requires repeated purification to achieve the desired chiral purity.
Another alternate route for the preparation of dorzolamide hydrochloride is disclosed in United States patent no.7109353 which involves the use of sodium perborate as an oxidant, as depicted in Scheme 4.
Scheme 4
chlorinating agent, cyclinization



Vl IV

VIl VlIl IX
The process disclosed in Scheme 4 has following disadvantages (a) Conversion of (i) to (ii) requires the mixture to be refluxed for 18-20 hrs which is time consuming and may cause impurity in the product.
(b) As the process uses the Ritter reaction to convert (vi) to (vii), a large amount of water is required to quench the hot mass of reaction which is not practical in an industrial set-up. (c) Sodium perborate is used as an oxidizing agent to convert (v) to (vi), which has got bleaching properties, and the handling of it may be injurious when done so for a prolonged period.
Yet another process for the preparation of dorzolamide is disclosed in United States publication no. 20060155132 which involves protecting the chiral 5,6-dihydro-4-(R)- hydroxy-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-7,7-dioxide as depicted in Scheme 5.
Scheme 5
protected amination benzyl sulphonyl chloride



The process disclosed in Scheme 5 has the following disadvantages, (a) The conversion process of compound (II) to (III) requires a very low temperature which ranges from -30° to 00C. (b) The amination process requires 16- 20 hrs, which is time consuming and may cause impurity in the product. All these disadvantages of the prior art are overcome by the process in accordance with the present invention.
Scheme 8

Example 4
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide
A suspension of 5,6-dihydro-4H-4-acetylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide (83.25 gms, 0.24 moles) in THF (832 ml) was cooled to 00C and sodium borohydride (49.11 gms, 1.29 moles) was added in lots maintaining temperature below 5°C. Reaction mass was stirred for 15 minutes at 5°C and boron trifluoride diethyl- etherate (249.75 ml, 287.2 gms, 2.02 moles) was added below 5°C. The reaction mass was stirred for 5 hours at 0°C to 5°C. Temperature of the reaction mass was raised to 25°C to 300C and stirred for 18 hours. The reaction mass was quenched in 1M sulphuric acid solution (1082 ml) below 5°C, temperature raised to 25°C to 30°C and stirred for 1 hour. The solvent was distilled under reduced pressure at 800C. The reaction mass was cooled to 100C and p H adjusted to 7 – 8 using 50% sodium hydroxide solution. Material was extracted in 1665 ml ethyl acetate once and 832 ml twice. The combined organic layers were washed with saturated sodium chloride solution, dried over sodium sulphate, charcoalised, filtered on hyflo, distilled to get title compound (77.42 gms). HPLC: 80:20::Trans:Cis
Example 7
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide-7,7-dioxide hydrochloride
(a) Dorzolamide di-p-toluyl-L-tartrate salt as prepared in example 6 (44.26 gms, 0.085 moles) was taken in ethyl acetate (557.0 ml), basified with saturated sodium bicarbonate solution. Reaction mass was stirred for 15 minutes at 25°C to 3O0C and aqueous layer was extracted with ethyl acetate (278 ml X 2). The organic layers were combined, washed with brine solution, dried over sodium sulphate, and charcoalized. To the clear solution, IPA + HCL (16.35 ml, 0.089 moles) was added, stirred for 30 minutes and ethyl acetate was removed by distillation at atmospheric pressure at 85°C to about 280 ml volume, cooled to 25-3O0C, stirred for 12 hours at same temperature and filtered to get 26.0 gms of dorzolamide hydrochloride. Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
(b) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(b).
(c) Dorzolamide hydrochloride was obtained in a similar manner in quantitative yield from the salt of example 6(c).
Example 8
Preparation of 5,6-Dihydro-4H-4-ethylamino-6-methylthieno[2,3-b]thiopyran-2- sulfonamide -7,7-dioxide hydrochloride without isolation of base
Dorzolamide di-p-toluyl-L-tartrate (50 gms, 0.096 moles) prepared as per example 6, was charged in a round bottom flask along with isopropanol (1000 ml). The reaction mass was heated to 800C and charged with IPA-HCI (20 ml) dropwise to pH 3 to 4. The reaction mass was heated to reflux for 5-10 minutes. The clear solution obtained was concentrated to 100 ml. The reaction mass was charged with 300 ml ethyl acetate, cooled to 25°C, stirred for 12 to 14 hours at same temperature. The resulting dorzolamide hydrochloride was isolated by filtration and washed with ethyl acetate (50 ml), dried under vacuum at 60- 65 0C for 5-6 hours. Yield- 30 gms.
Trans (-) dorzolamide hydrochloride > 99.5% Trans (+) dorzolamide hydrochloride < 0.5% Cis Isomer <0.1%
…………………………………………………………..

………………………………………
Dorzolamide hydrochloride, known chemically as 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran-2-sulfonamide-7,7-dioxyde hydrochloride, is a topically effective carbonic anhydrase inhibitor useful in the treatment of ocular hypertension.
Dorzolamide hydrochloride has the structure of Formula I:

U.S. Pat. Nos. 4,677,155 and 4,797,413 disclose Dorzolamide. In the prior art synthesis of dorzolamide, a chiral hydroxysulfone is used as a starting material. The chiral hydroxysulfone starting material can be obtained using the processes disclosed in U.S. Pat. Nos. 5,157,129, 5,474,919, and 5,760,249. In the disclosed processes, the chiral hydroxysulfone is obtained by the asymmetric enzymatic reduction of the corresponding ketosulfone, or by cyclization of the chiral thienyl thiobutyric acid, obtained, in turn, from a chiral hydroxyester or lactone, and the subsequent stereospecific reduction of the resulting ketone.
Processes for the preparation of dorzolamide hydrochloride are described in U.S. Pat. Nos. 4,797,413, 5,157,129, and 5,688,968 and in U.S. patent application Publication Ser. No. 2003/0220509. The disclosed processes involve conversion of a hydroxysulfone to the corresponding acetamidosulfone by a Ritter reaction with retention of configuration, followed by introduction of a sulfonamido group, and the subsequent reduction of the amido group to an amine, providing the desired product.
The process disclosed in U.S. Pat. No. 4,797,413 includes activation of the 4-hydoxy group of the sulfonaminated hydroxysulfone with tosyl chloride and the introduction of the desired alkylamino group by nucleophilic substitution, resulting in all diastereomeric products, which must be separated and resolved. As a result, at least 75 percent of the product is lost when the desired product is the more active enantiomer.

EXAMPLE 2
Preparation of 5,6-dihydro-4-(S)-ethylamino-6-(S)-methyl-4H-thieno-[2,3-b]thiopyran 7,7-dioxide hydrochloride salt (Formula IV)
Tetrahydrofuran (50 l) and triethyl amine (4.8 l) are added to 4-(R)-hydroxy-5,6-dihydro-6-(S)-methyl-4H-thieno[2,3b]thiopyran-7,7-dioxide (5 Kg) and stirred under a nitrogen atmosphere at room temperature. The solution is cooled to −10° C. Benzylsulfonyl chloride (5.4 Kg) solved in THF (15 l) is added to the DRZ-19 THF solution in portions while maintaining the temperature below 0° C. The feeding funnel is washed with THF (2 l). The reaction mixture is stirred at 0° C. for 2-4 hours. The formed TEA HCl is filtered and the cake is washed with THF (2×10 l) Ethylamine in THF (30%, 63.7 l) is added to the filtrate and the reaction mixture is stirred at 20°-25° C. for 16 hours. Ethylamine gas prepared by heating of 70% EtNH2water solution (50 l) is absorbed in cooled THF (30 l). Water (20 l) is added to the reaction mixture and THF is evaporated from the filtrate at 40°±5° C. under vacuum. The residue is cooled to 20°-25° C., ethyl acetate (60 l) is added to it and stirred vigorously. After phase separation, the organic phase is washed with water (20 l). The ethyl acetate phase is heated to 40°±2° C. and hydrochloric acid (4M, ˜8-10 l) is added to it during stirring to set pH 2.0-2.5. The formed slurry is cooled to −8°±2° C. and stirred for 3 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (30 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give the desired salt (˜5 Kg).
Preparation of dorzolamide hydrochloride product from the aminated intermediate of Formula IV
Fuming sulfuric acid (20%, 5 l) is cooled to −7°±2° C. and the aminated intermediate of Formula IV (2.5 Kg) is added to it in portions during stirring. The temperature of the reaction mixture is increased to 20°±5° C. during addition of the aminated intermediate of Formula IV. The reaction mixture is stirred for 22 hours at 20°±5° C. Thionyl chloride (20 l) is added to the stirred reaction mixture at 20°±5° C. The reaction mixture is heated to 60°-65° C. and stirred for 24 hours at this temperature. The mixture is cooled back to 40°±2° C. and the excess amount of thionyl chloride is evaporated at this temperature under vacuum. (The volume of the residue: ˜9 l.) The residue is cooled to −5°±2° C.
Ethyl acetate (75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature. The temperature of the diluted solution: 10°-25° C. Aqueous ammonia (25%, 75 l) is cooled to −10°±5° C. and the residue is added to it at this temperature during effective stirring, while maintaining the temperature below 30° C. The final pH: ˜11. The slurry is cooled to 0°±2° C. and stirred for 14 hours at this temperature. The formed ammonium sulfate is filtered and the cake is washed with ethyl acetate (2×20 l and 10 l ). Ethyl acetate is evaporated from the filtrate at 38°±2° C. under vacuum. The residue is heated to 38°±2° C., washed with toluene (3×37.5 l) at this temperature. Water (25 l) is added to the aqueous phase, cooled to 20°-25° C. and extracted with ethyl acetate (3×75 l, 37.5 l, and 37.5 l). The collected ethyl acetate phase is concentrated to ˜100 l at 38°±2° C. under vacuum. The residue is cooled to 20°-25° C. and hydrogen chloride in ethanol (5%, 10.8 l) is added to it during stirring. The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×20 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give Dorzolamide hydrochloride salt (˜2 Kg).
Crude Dorzolamide hydrochloride salt (9 Kg) is solved in water (225 l) at 20°-25° C. and the pH is set to 8.0-8.5 by addition of 25% of aqueous ammonia (2 l). The formed slurry is extracted with ethyl acetate (5×72 l). The collected ethyl acetate phase is concentrated to 180 l by vacuum distillation. The residue is cooled to 20°-25° C., ethyl acetate (45 l) and hydrogen chloride in ethanol (5%, 22.5 l) are added to it during stirring (pH:˜1.0). The formed slurry is stirred for 1 hour at 20°-25° C. then cooled to 0°-4° C. and stirred for 5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with ethyl acetate (2×30 l), and dried at 55°-60° C. under vacuum for 4-8 hours to give purified Dorzolamide hydrochloride salt (˜8.2 Kg).
Purified Dorzolamide hydrochloride salt (8 Kg) dissolved in water (24 l) at 95°-105° C. and treated with active carbon (80 g). After filtration, the water solution is cooled gradually to 0°-4° C. and stirred for 3-5 hours at this temperature. The slurry is filtered, the precipitated HCl salt is washed with cooled water (2×5 l) and dried at 55°-60° C. under vacuum for 4-8 hours to give crystallized DRZ HCl salt (˜6.6 Kg).
…………………………………………………………

Reaction of (I) with acetic anhydride-sulfuric acid in methylene chloride provided the sulfonic acid in 98% yield. Conversion to the sulfonyl chloride with phosphorous pentachloride in methylene chloride followed by treatment with aqueous ammonia gave the sulfonamide (II). Reduction of the carbonyl function with sodium borohydride and oxidation of the thiopyran sulfur with Oxone(R) yielded (IV). The 4-hydroxy substituent was converted to the acetylamino functionality under Ritter conditions. Reduction of (V) with borane-dimethylsulfide complex yielded (VI) as a mixture of diasteriomers. Chromatography on silica gel gave the trans-racemate, which was resolved into its individual enantiomers through the di-p-toluoyl-L-tartaric acid salt. The absolute configuration of the S,S-enantiomer, MK-507, was established by single crystal X-ray analysis.![]()

……………………………………………………….

………………………..

//////////A new synthesis of MK-0507 has been described: The condensation of 3(R)-(tosyloxy)butyric acid methyl ester (I) with lithium 2-thienylmercaptide (II) in formamide-THF gives 3(S)-(2-thienylthio)butyric acid methyl ester (III), which is hydrolyzed with aqueous HCl to the corresponding free acid (IV). The intramolecular Friedel-Crafts’cyclization of (IV) with trifluoroacetic anhydride yields 6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-one (V), which is reduced with LiAlH4 in toluene to afford 4(R)-hydroxy-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran (VI). Epimerization of (VI) with sulfuric acid gives the alcohol (VII) in a cis:trans ratio of 24:76%. Oxidation of (VII) with H2O2 and sodium tungstate yields the 7,7-dioxide (VIII; cis-trans mixture), which is acetylated with acetic anhydride to the acetate (IX). The reaction of (IX) with acetonitrile and sulfuric acid affords N-[6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-4-yl]acetamide 7,7-dioxide (X; cis-trans mixture), which is sulfonated with chlorosulfonic acid and then treated with SOCl2 to give 4-acetamide-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonyl chloride 7,7-dioxide (XI; cis-trans mixture). The reaction of (XI) with concentrated aqueous NH4OH in THF yields the corresponding sulfonamide (XII), which by reduction with BH3-dimethylsulfide in THF affords 4-(ethylamino)-6(S)-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide (XIII; cis-trans mixture). Finally, this mixture is treated with maleic acid in acetone and the resulting maleates are submitted to fractionated crystallization, giving the maleate of the (4S,6S)-isomer, which is treated first with NaHCO3 and then with HCl to give MK-0507; [alpha](25)589 -17.1 C (c 1, H2O).
H-NMR spectral analysis
 CAS NO. 130693-82-2, DORZOLAMIDE HCL H-NMR spectral analysis |
C-NMR spectral analysis
 CAS NO. 130693-82-2, DORZOLAMIDE HCL C-NMR spectral analysis |
References
- Dorzolamide at Drugs.com. Revised: 12/2011
- KD Tripari MD. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
Further reading
- Kubinyi H (1999). “Chance favors the prepared mind–from serendipity to rational drug design”. J Recept Signal Transduct Res19 (1–4): 15–39.doi:10.3109/10799899909036635. PMID 10071748.
- Plummer C, MacKay E, Gelatt K (2006). “Comparison of the effects of topical administration of a fixed combination of dorzolamide-timolol to monotherapy with timolol or dorzolamide on IOP, pupil size, and heart rate in glaucomatous dogs”. Vet Ophthalmol 9 (4): 245–9.doi:10.1111/j.1463-5224.2006.00469.x. PMID 16771760.
- Grover S, Apushkin M, Fishman G (2006). “Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa”.Am J Ophthalmol 141 (5): 850–8. doi:10.1016/j.ajo.2005.12.030.PMID 16546110.
- Almeida G, Faria e Souza S (2006). “Effect of topical dorzolamide on rabbit central corneal thickness”. Braz J Med Biol Res 39(2): 277–81.doi:10.1590/S0100-879X2006000200015. PMID 16470316.
Reference:
CIPLA LIMITED; CURTIS, Philip, Anthony Patent: WO2008/135770 A2, 2008 ; Location in patent: Page/Page column 21-22 ;
RAGACTIVES, S.L. Patent: US2003/220509 A1, 2003 ; Location in patent: Page/Page column 12 ;
WO2011/101704 A1, ;
Literature References:
Carbonic anhydrase inhibitor. Prepn: J. J. Baldwin et al., EP 296879; eidem,US 4797413 (1988, 1989 both to Merck & Co.). Mechanism of action study: R.-F. Wang et al., Arch. Ophthalmol. 109, 1297 (1991).
HPLC determn in plasma and urine: B. K. Matuszewski, M. L. Constanzer,Chirality 4, 515 (1992).
Clinical evaluations in glaucoma and ocular hypertension: E. A. Lippa et al.,Ophthalmology 98, 308 (1991); E. A. Lippa et al., Arch. Ophthalmol. 110, 495 (1992).
| REFERENCE | ||
|---|---|---|
| 1 | * | KAMEI K. ET AL.: ‘Chemical structure, physico-chemical properties and stability of dorzolamide hydrochloride‘ IYAKUHIN KENKYU vol. 25, no. 6, 1994, pages 438 – 452, XP008040715 |
| 2 | * | QUINT M.-P. ET AL.: ‘Dorsolamide hydrochloride‘ ANALYTICAL PROFILES OF DRUG SUBSTANCES AND EXCIPIENTS vol. 26, 1999, pages 283 – 316, XP008040718 |
| EP2128161A1 * | May 30, 2008 | Dec 2, 2009 | Ragactives, S.L. | Process for obtaining 4-hydroxy-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-7,7-dioxide and its enantiomers, and applications thereof |
| WO2008135770A2* | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |
| WO2009144263A2* | May 28, 2009 | Dec 3, 2009 | Ragactives, S.L.U. | PROCESS FOR OBTAINING 4-HYDROXY-6-METHYL-5, 6-DIHYDRO-4H-THIENO [2,3-b] THIOPYRAN-7, 7-DIOXIDE AND ITS ENANTIOMERS, AND APPLICATIONS THEREOF |
| WO2014005943A1* | Jun 28, 2013 | Jan 9, 2014 | Zach System S.P.A. | Process for preparing enantiomerically enriched oxamides |
| US8263787 | May 7, 2008 | Sep 11, 2012 | Cipla Limited | Process for preparing dorzolamide |
| WO1994021645A1* | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| EP0296879A1 * | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| US5474919 * | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5760249 * | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US20060142595 * | Dec 28, 2004 | Jun 29, 2006 | Council Of Scientific & Industrial Research | Starting by reacting a 2-halothiophene with a Grignard reagent in a solvent in situ with sulfur, triethylamine hydrochloride, crotonic acid and a base; product is chlorinated, cyclized, chlorosulfonated and aminated, reduced, oxidized, amidated, hydrogenated, neutralized, recrystallized and resolved |
| US5157129 | Apr 18, 1990 | Oct 20, 1992 | Merck & Co., Inc. | Enantiospecific synthesis of s-(+)-5,6-dihydro-4-(r-amino)-4h-thieno(2,3-b)thiopyran-2-sulfonamide-7,7-dioxide |
| US5474919 | Sep 13, 1994 | Dec 12, 1995 | Merck & Co., Inc. | Bioconversion process for the synthesis of transhydroxy sulfone by Rhodotorula rubra or Rhodotorula piliminae |
| US5688968 | Jan 6, 1995 | Nov 18, 1997 | Merck & Co., Inc. | Enantioselective synthesis of 5,6-dihydro-(S)-4-(ethylamino)-(S)-6-methyl-4H-thieno 2,3-B!thiopyran-2-sulfonamide 7,7-dioxide |
| US5760249 | Aug 28, 1996 | Jun 2, 1998 | Merck & Co., Inc. | Synthesis of hydroxysulfone and related compounds |
| US7109353 | Dec 28, 2004 | Sep 19, 2006 | Council Of Scientific And Industrial Research | Process for preparing 5,6-dihydro-4-(S)-(ethylamino)-6-(S) methyl-4H-thieno[2,3b]thiopyran-2-sulphonamide-7,7-dioxide HCl |
| US20060155132 | Jan 6, 2006 | Jul 13, 2006 | Kovacs Laszlo Z | Method of making dorzolamide hydrochloride |
| EP0296879A1 | Jun 23, 1988 | Dec 28, 1988 | Merck & Co., Inc. | Substituted aromatic sulfonamides as antiglaucoma agents |
| WO1994021645A1 | Mar 16, 1994 | Sep 29, 1994 | Thomas J Blacklock | ENANTIOSELECTIVE SYNTHESIS OF 5,6-DIHYDRO-(S)-4-(ETHYLAMINO)-(S)-6-METHYL-4H-THIENO[2,3-b]THIOPYRAN-2-SULFONAMIDE 7,7-DIOXIDE AND RELATED COMPOUNDS |
| WO2008135770A2 | May 7, 2008 | Nov 13, 2008 | Cipla Ltd | Process for preparing dorzolam ide |
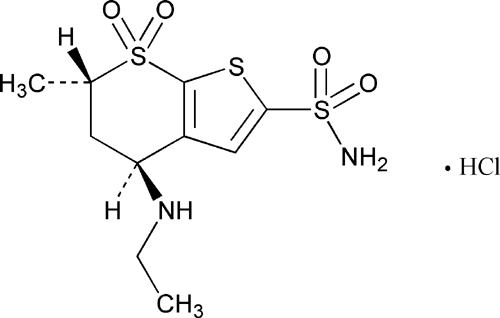
↧
9 Likely Signs you have a Hormone Imbalance

9 Likely Signs you have a Hormone Imbalance
Being able to spot the signs you have a Hormone imbalance is key to maintaining a holistic health lifestyle because they play such an important role in virtually every aspect your well being.
What do your Hormones do?
Your hormones are tiny little chemical messengers that whizz around your bloodstream telling your organs and body tissues to fulfill their functions. These busy little chaps affect your well being in various ways, which include:
- Your development and growth
- The way you metabolize food
- Sexual functionality
- Cognition and mood
- Body temperature
As you can see they impact on all aspects of your health and well-being. So making sure that you keep them in balance is crucial to your holistic health lifestyle. Unfortunately, it doesn't take much to throw them out of balance, so we're going to be looking at nine tell-tale signs that will warn you that all is not quite right on the farm - that farm being your body of course.
The most publicized hormonal changes...
http://eatlocalgrown.com/article/12410-9-likely-signs-you-have-a-hormone-imbalance.html?c=nsm
ABOUT THE AUTHOR

Michelle Toole
Michelle Toole runs a popular health related website, Healthy Holistic Living.com. Michelle is an independent health researcher or, to put it simply, an information seeker. Michelle would describe herself as an inventor, entrepreneur, mother, adventurer, author and webmaster.
You can find more health related information and tips on Michelle’s website healthyholisticliving.com or on her popular
Michelle Toole runs a popular health related website, Healthy Holistic Living.com.
Michelle is an independent health researcher or, to put it simply, an information seeker.
Michelle would describe herself as an inventor, entrepreneur, mother, adventurer, author and webmaster.
You can find more health related information and tips
You can find more health related information and tips
on Michelle’s website healthyholisticliving.com or on her popular



more info from other sources
Hormones work as chemical messengers in our body. We go through hormonal changes from birth to death. They cause changes in our bodies over time including:
- Reproduction
- Growth & Development
- Metabolism
- Mood
- Sexual Function

Knowing if you are suffering from hormonal imbalance is important. Here are some of the most common signs of hormonal imbalance and ways to fix it:
1- Gaining Weight Persistently
If you are gaining weight consistently despite a healthy diet and appropriate physical activity it can be a sign of hormonal imbalance. It can be due to unaddressed or emerging insulin resistance. Somethyroid problems can also cause weight gain. You need to see your doctor for further evaluation. In the meantime, it’s highly recommended to avoid processed foods, sugar (soda), and wheat.
2- Insomnia
If you are suffering from insomnia and having difficulty sleeping it can be a sign of hormonal imbalance. Try to optimize your sleep and take antioxidants. Make sure to include quality lean protein, especially at dinner. Try exercise and yoga to improve your sleep. Also avoid these common habits that may cause insomnia.
3- Chronic Stress
Chronic stress depletes adrenal functions which leads to low levels of progesterone and other important hormones. Try to work on becoming stress-free. A healthy diet and changing your environment can help you reduce stress.
4- Excessive Sweating
If you are have night sweats and hot flashes it can be due to hormonal imbalance. Write down your routine, what you eat and drink, how you feel, and what kind of emotions cause your temperature to rise. Work on these areas next time when you get hot flashes.
5- Cravings
Eating abnormally or feeling that you still want to have more even after eating can be an indicator that something is amiss. It’s due to hormonal imbalance like adrenal fatigue or insulin resistance. Eliminating sugars, dairy/ wheat products and alcohol will help you control your cravings and improve your digestion. To learn more about how to improve digestion
6- Depression
Feeling depressed and rejected is a sign of hormonal imbalance. If not clinically caused it can mean you’re not feeding your body what it needs. Listen to your inner self and treat your body in a good way with a healthy diet, exercise and proper nutrients. You may want to consult with a mental health professional for further evaluation.
7- Fatigue
Feeling tired by the end of first half? Or feeling sluggish while working? Hormonal imbalance could be the reason for your fatigue. A proper diet enriched with nutrients and eliminating wheat and grains may help stabilize your blood sugar. It’s very common that gluten-intolerance causes fatigue. Here is a guide to fix fatigue.
8- Low Libido
This is one of the most noticeable signs of hormonal imbalance. Often imbalance in sex hormones, thyroid or other endocrine glands is the underlying cause of low sex drive. It also happens due to lack of sound sleep. Work on your sleeping habits and optimize your sleep.
9- Digestive Problems
Slow digestion and gastric problems are also among common symptoms of hormonal imbalance. Hormonal problems during perimenopause is one of the most common causes of digestive problems for women between the ages of 45 and 55. This is a useful guide to improve digestion naturally.

↧
Ayurveda………..Medicinal Benefits of Liquorice (Mulethi) (मुलेठी, 甘草, شیرین بیان)
Ayurveda………..Medicinal Benefits of Liquorice (Mulethi) (मुलेठी, 甘草, شیرین بیان)

Licorice or Mulethi is a medicinal herb which is used in various Ayurvedic medicines. Its underground stems and roots are used for medicinal purpose. It has antioxidant, antimicrobial, anti-inflammatory and hepatoprotective properties.
Mulethi is useful in cough, sore throat, bronchitis, sexual weakness, skin problems, jaundice, hoarseness, vata dosha, ulcers etc. It has demulcent and expectorant properties.
Mulethi is useful in cough, sore throat, bronchitis, sexual weakness, skin problems, jaundice, hoarseness, vata dosha, ulcers etc. It has demulcent and expectorant properties.
read…………MY OLD ARTICLE

Liquorice, or licorice, (/ˈlɪk(ə)rɪʃ/ lik-(ə-)rish or /ˈlɪk(ə)rɪs/ lik-(ə-)ris)[2] is the root ofGlycyrrhiza glabra from which a sweet flavour can be extracted. The liquorice plant is alegume native to southern Europe, India, and parts of Asia. It is not botanically related toanise, star anise, or fennel, which are sources of similar flavouring compounds. The word liquorice / licorice is derived (via the Old French licoresse) from the Greek γλυκύρριζα (glukurrhiza), meaning “sweet root”,[3] from γλυκύς (glukus), “sweet”[4] + ῥίζα (rhiza), “root”,[5][6] the name provided by Dioscorides.[7] It has been traditionally known and used as medicine in Ayurveda for rejuvenation.[8] It is called asadhimadhuram (அதிமதுரம்) in Tamil, irattimadhuram in Malayalam, yastimadhu (यस्टिमधु) in Sanskrit, mulethi (मुलेठी) in Hindi, andjethimadh (જેઠીમધ) in Gujarati language.[9]
Licorice (Glycyrrhiza glabra), locally known as mulethi, has been revered for centuries as a medicinal herb in Ayurveda. Besides possessing numerous medicinal properties, it is also a popular flavoring herb as it is 50 times sweeter than sugar, due to the presence of a compound called glycyrrhizin.
Through research, the anti-oxidant, anti-inflammatory, anti-microbial, analgesic (pain-relieving) and expectorant properties of this is sweet, moist herb have been established worldwide. It is also diuretic, rejuvenating and mildly laxative in nature. These properties have helped Licorice find a place in both Eastern and Western medicine for treating an array of ailments, ranging from cold and cough to arthritis, respiratory, digestive and liver problems.

The Sanskrit name for licorice is Yashtimadhu, which literally means “sweet root”. It is sweet, cool and heavy to digest. The Rasa (taste) of this herb is madhura (sweet), which makes it beneficial for vata and pitta doshas, while it’s Virya (action) is sheetal (cooling), which generally increases kapha when consumed in large doses over long term.
The medicinal property of mulethi is mainly because of the presence of powerful phytochemicals namely flavonoids, chalcones, saponins and xenoestrogens. Glycyrrhizin (salts of glycyrrhizic acid) is a popular saponin found in roots of mulethi that is responsible for the characteristic sweet taste (50 times more sweet than sugar) flavor. Liquiritin, licoflavonol, liquiritigenin, etc are the common chalcones that provide the distinct yellowish color to mulethi; while, the aroma of its root is mainly because of anethole. Here are the ten health benefits of mulethi:

Information
Latin name: Glycyrrhiza glabra
Sanskrit: Madhuyashti
Hindi: Mulhatti, Jethimadh, Mithilakdi
English: Sweetwood, Liquorice, Licorice
Bengali: Jashtimadhu
Gujrati: Jethi Madh
Marathi: Jeshtamadhu
Kannada: Jeshthamadhu
Malayalam: Itarttimadhuram, Erattimadhuram
Tamil: Atimadhuram
Telugu: Atimadhuramu
Sanskrit: Madhuyashti
Hindi: Mulhatti, Jethimadh, Mithilakdi
English: Sweetwood, Liquorice, Licorice
Bengali: Jashtimadhu
Gujrati: Jethi Madh
Marathi: Jeshtamadhu
Kannada: Jeshthamadhu
Malayalam: Itarttimadhuram, Erattimadhuram
Tamil: Atimadhuram
Telugu: Atimadhuramu

Anti-microbial activity – Roots of mulethi are very effective in protecting against virus, bacteria and fungi due to the presence of Glycyrrhizin that blocks the microbial growth. The root extract possesses the power to control malaria (as per preliminary research), influenza and also helps in the treatment of herpes resulting in virus suppression and severity of sores.
Anti-inflammatory activity – Liquorice has powerful anti-inflammatory and anti-allergic activity and can be used to treat chronic inflammation like rheumatic problems & arthritis, skin diseases and autoimmune diseases. It is also used for preventing any inflammatory conditions related to eye and also to treat conjunctivitis with the help of glycyrrhizin activity that counteracts negative effects caused by cortisol.
Improves immunity – Root extracts of mulethi aids in increasing the production of lymphocytes and macrophage thereby improving your defense mechanism & preventing microbial attack. It also helps in minimizing immune related allergic reactions and autoimmune complications.

Memory improvement – Roots of licorice exert supportive effect on the adrenal gland and thus indirectly aid in stimulating the brain. It not only decreases the effects of amnesia & improves learning but its antioxidant property (mulethi contains flavonoids) renders a shielding effect on the brain cells.
Anti-ulcer activity – The potent antioxidant and anti-inflamatory properties of licorice makes it the best natural medicinal aid to treat ulcers of stomach, intestine and mouth. The compound carbenoxolone synthesized from glycyrrhizin plays key role in healing mouth and gastric ulcers along with reducing gastric secretions and promoting development of intestinal mucus lining.
Liver protection – Licorice is one of the most common traditional remedy used to treat jaundice. Its antioxidant property is the key for preventing your liver from the action of free radicals and toxic materials. This herb is also reported to exhibit protection against diclofenac induced toxicity and also, in inhibiting damage of liver.

Digestive aid – Roots of licorice are also used to deal with stomach and digestion problems with the help of glycyrrhizin and its compound, carbenoxolone. It is one of the ancient home remedies for relieving constipation, acidity, heartburn, stomach discomfort, inflammation of digestive system and gastro esophageal acid reflux. As a mild laxative, it plays an effective role in bowel movements and also for treatment of allergic cough in addition to maintaining normal pH levels.
Hormonal regulation – The phytoestrogenic compounds present in mulethi roots exert valuable action against women hormonal imbalance problems, menopause symptoms like hot flashes & exhaustion, mood swings, etc. It is also found to help in cortisol production and relieving premenstrual issues like nausea and menstrual cramps. Licorice powder acts as the traditional medicine for nursing mothers to regulate body hormones and aid in milk secretion.
Heart healthy effects – Research studies have proved that licorice roots help in controlling cholesterol levels by increasing the body’s flow of bile and also reducing high blood cholesterol levels. The anti-oxidant property of licorice acts in increasing the blood capillary health, reducing inflammation, prevents blood vessel damage and block development of arterial plaque.
Other effects – Licorice roots work wonders in treatment of depression, diabetes and respiratory tract infection like sore throat (hoarseness of voice), cold and cough, etc in addition to rendering effective skin benefits, oral hygiene and weight loss. It is found to act as a cancer cure remedy, a potent aphrodisiac and a powerful analgesic agent.
Description
It is a herbaceous perennial, growing to 1 m in height, with pinnate leaves about 7–15 cm (3–6 in) long, with 9–17 leaflets. The flowers are 0.8–1.2 cm (1/3 to 1/2 in) long, purple to pale whitish blue, produced in a loose inflorescence. The fruit is an oblong pod, 2–3 cm (1 in) long, containing several seeds.[10] The roots are stoloniferous.[11]
Chemistry
The scent of liquorice root comes from a complex and variable combination of compounds, of which anethole is up to 3% of total volatiles. Much of the sweetness in liquorice comes from glycyrrhizin, which has a sweet taste, 30–50 times the sweetness of sugar. The sweetness is very different from sugar, being less instant, tart, and lasting longer.
The isoflavene glabrene and the isoflavane glabridin, found in the roots of liquorice, arephytoestrogens.[12][13]
Cultivation and uses
Liquorice, which grows best in well-drained soils in deep valleys with full sun, is harvested in the autumn two to three years after planting.[10] Countries producing liquorice include Iran, Afghanistan, the People’s Republic of China, Pakistan, Iraq, Azerbaijan, Uzbekistan, Turkmenistan, and Turkey.[14]
The world’s leading manufacturer of liquorice products is M&F Worldwide, which manufactures more than 70% of the worldwide liquorice flavours sold to end users.[15]
Safe dosage
Licorice is available in various forms – root, powder and extracts. Licorice root can be chewed directly while licorice tea (prepared by boiling licorice root in water) is also extremely beneficial as a home remedy.
Daily intake of 5-6 grams of licorice powder is considered safe while 250-500 mg of concentrated extracts can be taken thrice a day. Unsupervised use in high doses is not recommended for long term. People with hypertension or heart disease, pregnant women and breastfeeding mothers should avoid using licorice without prior consulation with an Ayurveda doctor.
 plant
plant
Medicine
The compound glycyrrhizin (or glycyrrhizic acid), found in liquorice, has been proposed as being useful for liver protection in tuberculosis therapy, but evidence does not support this use, which may in fact be harmful.[24] Glycyrrhizin has also demonstrated antiviral, antimicrobial, anti-inflammatory, hepatoprotective, and blood pressure-increasing effects in vitro and in vivo, as is supported by the finding that intravenous glycyrrhizin (as if it is given orally very little of the original drug makes it into circulation) slows the progression of viral and autoimmune hepatitis.[25][26] Liquorice has also demonstrated promising activity in one clinical trial, when applied topically, against atopic dermatitis.[27] Additionally, liquorice has also proven itself effective in treating hyperlipidaemia (a high amount of fats in the blood).[28]Liquorice has also demonstrated efficacy in treating inflammation-induced skinhyperpigmentation.[29][30] Liquorice may also be useful in preventing neurodegenerative disorders and dental caries.[31][32][33]
The antiulcer, laxative, antidiabetic, anti-inflammatory, immunomodulatory, antitumour andexpectorant properties of liquorice have been investigated.[34]
Folk medicine
In traditional Chinese medicine, liquorice (मुलेठी, 甘草, شیرین بیان) is believed to “harmonize” the ingredients in a formula and to carry the formula to the 12 “regular meridians”.[35]
References
- “Glycyrrhiza glabra information from NPGS/GRIN”. http://www.ars-grin.gov. Retrieved6 March 2008.
- licorice. Merriam-Webster’s Medical Dictionary, © 2007 Merriam-Webster, Inc.
- γλυκύρριζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- γλυκύς, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus
- Jump up^ ῥίζα, Henry George Liddell, Robert Scott, A Greek-English Lexicon, on Perseus<
- Jump up^ liquorice, on Oxford Dictionaries
- Jump up^ google books Maud Grieve, Manya Marshall – A modern herbal: the medicinal, culinary, cosmetic and economic properties, cultivation and folk-lore of herbs, grasses, fungi, shrubs, & trees with all their modern scientific uses, Volume 2 Dover Publications, 1982 & Pharmacist’s Guide to Medicinal Herbs Arthur M. Presser Smart Publications, 1 Apr 2001 2012-05-19
- Jump up^ Balakrishna, Acharya (2006). Ayurveda: Its Principles & Philosophies. New Delhi, India: Divya prakashan. p. 206. ISBN 8189235567.
- Jump up^ “Top 10 health benefits of Mulethi or Liquorice”.
- ^ Jump up to:a b Huxley, A., ed. (1992). New RHS Dictionary of Gardening. ISBN 0-333-47494-5
- Jump up^ Brown, D., ed. (1995). “The RHS encyclopedia of herbs and their uses”.ISBN 1-4053-0059-0
- Jump up^ Somjen, D.; Katzburg, S.; Vaya, J.; Kaye, A. M.; Hendel, D.; Posner, G. H.; Tamir, S. (2004). “Estrogenic activity of glabridin and glabrene from licorice roots on human osteoblasts and prepubertal rat skeletal tissues”. The Journal of Steroid Biochemistry and Molecular Biology 91 (4–5): 241–246.doi:10.1016/j.jsbmb.2004.04.008. PMID 15336701.
- Jump up^ Tamir, S.; Eizenberg, M.; Somjen, D.; Izrael, S.; Vaya, J. (2001). “Estrogen-like activity of glabrene and other constituents isolated from licorice root”. The Journal of steroid biochemistry and molecular biology 78 (3): 291–298. doi:10.1016/S0960-0760(01)00093-0. PMID 11595510.
- ^ Jump up to:a b c M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2010.
- Jump up^ M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2001.
- Jump up^ Erik Assadourian, Cigarette Production Drops, Vital Signs 2005, at 70.
- Jump up^ M & F Worldwide Corp., Annual Report on Form 10-K for the Year Ended December 31, 2005.
- ^ Jump up to:a b c Marvin K. Cook, The Use of Licorice and Other Flavoring Material in Tobacco (Apr. 10, 1975).
- Jump up^ Boeken v. Phillip Morris Inc., 127 Cal. App. 4th 1640, 1673, 26 Cal. Rptr. 3d 638, 664 (2005).
- Jump up^ [1] the online Dutch food composition database]
- Jump up^ “Right good food from the Ridings”. AboutFood.com. 25 October 2007.
- Jump up^ “Where Liquorice Roots Go Deep”. Northern Echo. Retrieved 9 December2008.
- Jump up^ http://science.howstuffworks.com/life/botany/licorice-info.htm
- Jump up^ Liu Q, Garner P, Wang Y, Huang B, Smith H (2008). “Drugs and herbs given to prevent hepatotoxicity of tuberculosis therapy: systematic review of ingredients and evaluation studies”.BMC Public Health (Systematic review) 8: 365.doi:10.1186/1471-2458-8-365. PMC 2576232. PMID 18939987.
- Jump up^ Chien, CF; Wu, YT; Tsai, TH (January 2011). “Biological analysis of herbal medicines used for the treatment of liver diseases.”. Biomedical Chromatography 25(1-2): 21–38.doi:10.1002/bmc.1568. PMID 21204110.
- Jump up^ Yasui, S; Fujiwara, K; Tawada, A; Fukuda, Y; Nakano, M; Yokosuka, O (December 2011). “Efficacy of intravenous glycyrrhizin in the early stage of acute onset autoimmune hepatitis.”.Digestive Diseases and Sciences 56 (12): 3638–47.doi:10.1007/s10620-011-1789-5. PMID 21681505.
- Jump up^ Reuter, J; Merfort, I; Schempp, CM (2010). “Botanicals in dermatology: an evidence-based review.”. American Journal of Clinical Dermatology 11 (4): 247–67.doi:10.2165/11533220-000000000-00000. PMID 20509719.
- Jump up^ Hasani-Ranjbar, S; Nayebi, N; Moradi, L; Mehri, A; Larijani, B; Abdollahi, M (2010). “The efficacy and safety of herbal medicines used in the treatment of hyperlipidemia; a systematic review.”. Current pharmaceutical design 16 (26): 2935–47. doi:10.2174/138161210793176464. PMID 20858178.
- Jump up^ Callender, VD; St Surin-Lord, S; Davis, EC; Maclin, M (April 2011). “Postinflammatory hyperpigmentation: etiologic and therapeutic considerations.”.American Journal of Clinical Dermatology12 (2): 87–99. doi:10.2165/11536930-000000000-00000. PMID 21348540.
- Jump up^ Leyden, JJ; Shergill, B; Micali, G; Downie, J; Wallo, W (October 2011). “Natural options for the management of hyperpigmentation.”. Journal of the European Academy of Dermatology and Venereology 25 (10): 1140–5. doi:10.1111/j.1468-3083.2011.04130.x. PMID 21623927.
- Jump up^ Kannappan, R; Gupta, SC; Kim, JH; Reuter, S; Aggarwal, BB (October 2011). “Neuroprotection by spice-derived nutraceuticals: you are what you eat!”(PDF). Molecular Neurobiology 44(2): 142–59. doi:10.1007/s12035-011-8168-2.PMC 3183139. PMID 21360003.
- Jump up^ Gazzani, G; Daglia, M; Papetti, A (April 2012). “Food components with anticaries activity.”. Current Opinion in Biotechnology 23 (2): 153–9.doi:10.1016/j.copbio.2011.09.003.PMID 22030309.
- Jump up^ Messier, C; Epifano, F; Genovese, S; Grenier, D (January 2012). “Licorice and its potential beneficial effects in common oro-dental diseases.”. Oral Diseases 18(1): 32–9.doi:10.1111/j.1601-0825.2011.01842.x. PMID 21851508.
- Jump up^ Shibata, S (October 2000). “A drug over the millennia: pharmacognosy, chemistry, and pharmacology of licorice.”. Yakugaku Zasshi 120 (10): 849–62.PMID 11082698.
- Jump up^ Bensky, Dan; et al. (2004). Chinese Herbal Medicine: Materia Medica, Third Edition. Eastland Press. ISBN 0-939616-42-4.
- Jump up^ Olukoga, A; Donaldson, D (June 2000). “Liquorice and its health implications.”. The Journal of the Royal Society for the Promotion of Health 120 (2): 83–9.doi:10.1177/146642400012000203. PMID 10944880.
- Jump up^ Armanini, D; Fiore, C; Mattarello, MJ; Bielenberg, J; Palermo, M (September 2002). “History of the endocrine effects of licorice.”. Experimental and Clinical Endocrinology & diabetes 110 (6): 257–61. doi:10.1055/s-2002-34587.PMID 12373628.
- Jump up^ Omar, Hesham R; Komarova,, Irina; El-Ghonemi,, Mohamed; Ahmed, Fathy; Rashad, Rania; Abdelmalak, Hany D; Yerramadha, Muralidhar Reddy; Ali, Yaseen; Camporesi, Enrico M. “How much is too much? in Licorice abuse: time to send a warning message from Therapeutic Advances in Endocrinology and Metabolism”. http://www.ncbi.nlm.nih.gov. SAGE Publications. Retrieved 13 January2015.
38 Toxicology Center[2]
External links
↧
Logistics of process R&D: transforming laboratory methods to manufacturing scale
Logistics of process R&D: transforming laboratory methods to manufacturing scale

The manufacture of a | omeprazole (racemic product; top), and esomeprazole (the (S)-enantiomer; bottom), including b | a flow chart of the process for the …
Nature Reviews Drug Discovery 2, 654-664 (August 2003) | doi:10.1038/nrd1154
Logistics of process R&D: transforming laboratory methods to manufacturing scale
Hans-Jürgen Federsel
In the past, process R&D — which is responsible for producing candidate drugs in the required quantity and of the requisite quality — has had a low profile, and many people outside the field remain unaware of the challenges involved. However, in recent years, the increasing pressure to achieve shorter times to market, the demand for considerable quantities of candidate drugs early in development, and the higher structural complexity — and therefore greater cost — of the target compounds, have increased awareness of the importance of process R&D. Here, I discuss the role of process R&D, using a range of real-life examples, with the aim of facilitating integration with other parts of the drug discovery pipeline.
Process R&D, AstraZeneca, SE-151 85 Södertälje, Sweden. Hans-Jurgen.Federsel@astrazeneca.com
↧
↧
Novartis obtains European approval for Cosentyx to treat psoriasis
Novartis obtains European approval for Cosentyx to treat psoriasis

Novartis obtains European approval for Cosentyx to treat psoriasis
Swiss drug-maker Novartis has received approval from the European Commission (EC) for its Cosentyx (secukinumab, formerly known as AIN457) to treat moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy.SEE
Swiss drug-maker Novartis has received approval from the European Commission (EC) for its Cosentyx (secukinumab, formerly known as AIN457) to treat moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy.SEE
 PSORIAIS
PSORIAISsecukinumab

Secukinumab is a human monoclonal antibody designed for the treatments of uveitis,rheumatoid arthritis, ankylosing spondylitis, and psoriasis. It targets member A from thecytokine family of interleukin 17.[1][2] At present, Novartis Pharma AG, the drug’s developer, plans to market it under the trade name “Cosentyx.” [3] It is highly specific to the human immunoglobulin G1k (IgG1k) subclass.[2]
In July 2014 secukinumab established superiority to placebo and to etanercept for the treatment of chronic plaque psoriasis in Phase III clinical trials.[4] In October 2014, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee unanimously voted to recommend the drug for FDA approval, although this vote in and of itself does not constitute an approval. However, the FDA typically follows recommendations from these committees.[5] In October 2014, Novartis announced that the drug had achieved a primary clinical endpoint in two phase III clinical trials for ankylosing spondylitis.[6] As of 28 October, the relevant FDA committee had not yet responded to these results. In early November 2014, Novartis also released the results of a Phase 3 study on Psoriatic Arthritis that yielded very promising results.[7]

Although the drug was originally intended to treat rheumatoid arthritis, phase II clinical trials for this condition yielded disappointing results.[8] Similarly, while patients in a phase II clinical trial for [psoriatic arthritis] did show improvement over placebo, the improvement did not meet adequate endpoints and Novartis is considering whether to do more research for this condition.[9] Novartis has said that it is targeting approval and release in early 2015 for plaque psoriasis and ankyloding spondylitis indications.
It is also in a phase II clinical trial for Multiple Sclerosis [10] as it has exhibited efficacy in treating experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833.
- http://www.medscape.com/viewarticle/835331
- Langley RG, Elewski BE, Mark Lebwohl M, et al., for the ERASURE and FIXTURE Study Groups (July 24, 2014). “Secukinumab in Plaque Psoriasis — Results of Two Phase 3 Trials”. N Engl J Med 371: 326–338. doi:10.1056/NEJMoa1314258.
- committees.http://www.familypracticenews.com/index.php?id=2934&type=98&tx_ttnews=306073
- http://inpublic.globenewswire.com/2014/10/23/Novartis+AIN457+secukinumab+meets+primary+endpoint+in+two+Phase+III+studies+in+ankylosing+spondylitis+a+debilitating+joint+condition+of+the+spine+HUG1864939.html
- http://www.medpagetoday.com/MeetingCoverage/ACR/48743
- http://www.medscape.com/viewarticle/806510_6
- http://www.ncbi.nlm.nih.gov/pubmed/23361084
- http://clinicaltrials.gov/show/NCT01874340
| MONOCLONAL ANTIBODY | |
|---|---|
| TYPE | Whole antibody |
| SOURCE | Human |
| TARGET | IL17A |
| CLINICAL DATA | |
| LEGAL STATUS |
|
| IDENTIFIERS | |
| CAS NUMBER | |
| ATC CODE | L04AC10 |
| DRUGBANK | DB09029 |
| SYNONYMS | AIN457 |
| CHEMICAL DATA | |
| FORMULA | C6584H10134N1754O2042S44 |
| MOLECULAR MASS | 147.94 kDa |
↧
Rupatadine
Rupatadine

Rupatadine
CAS 158876-82-5,
8-Chloro-11-(1-((5-methylpyridin-3-yl)methyl)piperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine,
8-chloro-11-(1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
UNII-2AE8M83G3E, UR 12592
Molecular Formula: C26H26ClN3
Molecular Weight: 415.95774 g/mol
Percent Composition: C 75.07%, H 6.30%, Cl 8.52%, N 10.10%
Properties: Creamy solid, mp 58-61°.
Melting point: mp 58-61°
Platelet activating factor receptor antagonist; Histamine H1 receptor antagonist
Allergic rhinitis; Urticaria
| J. Uriach & Cia. S.A. |

Uriach developed and launched rupatadine for treating of allergic rhinitis and urticaria. Family members of the product case EP577957, have SPC protection in the EU until 2016.
As of January 2015, Newport Premium™ reports that Cadila Pharmaceuticals is producing or capable of producing commercial quantities of rupatadine fumarate and holds an active USDMF since September 2012.
Rupatadine is a second generation antihistamine and PAF antagonist used to treat allergies. It was discovered and developed by J. Uriach y Cia, S. A.[1] and is marketed under several trade names such as Rupafin, Alergoliber, Rinialer, Pafinur, Rupax and Ralif.
Therapeutic indications approved
Rupatadine fumarate has been approved for the treatment of allergic rhinitis and chronic urticaria in adults and children over 12 years. The defined daily dose (DDD) is 10 mg orally.

Derivative Type: Fumarate
CAS Registry Number: 182349-12-8
Trademarks: Rupafin (Uriach)
Molecular Formula: C26H26ClN3.C4H4O4
Molecular Weight: 532.03
Percent Composition: C 67.73%, H 5.68%, Cl 6.66%, N 7.90%, O 12.03%
Derivative Type: Trihydrochloride
CAS Registry Number: 156611-76-6
Molecular Formula: C26H26ClN3.3HCl
Molecular Weight: 525.34
Percent Composition: C 59.44%, H 5.56%, Cl 26.99%, N 8.00%
Properties: Crystals from ethyl acetate + ether, mp 213-217°.
Melting point: mp 213-217°.
Therap-Cat: Antihistaminic.
Keywords: Antihistaminic; Tricyclics; Other Tricyclics; Platelet Activating Factor Antagonist.
Available form
Rupatadine is available as round, light salmon coloured tablets containing 10 mg of rupatadine (as fumarate) to be administered orally, once a day.
Side effects
Rupatadine is a non-sedating antihistamine. However, as in other non sedating second-generation antihistamines, the most common side effects in controlled clinical studies weresomnolence, headaches and fatigue.
Mechanism of action
Rupatadine is a second generation, non-sedating, long-acting histamine antagonist with selective peripheral H1 receptor antagonist activity. It further blocks the receptors of the platelet-activating factor (PAF) according to in vitro and in vivo studies.[2]
Rupatadine possesses anti-allergic properties such as the inhibition of the degranulation ofmast cells induced by immunological and non-immunological stimuli, and inhibition of the release of cytokines, particularly of the TNF in human mast cells and monocytes.[3]

Pharmacokinetics
Rupatadine has several active metabolites such as desloratadine, 3-hydroxydesloratadine, 6-hydroxydesloratadine and 5-hydroxydesloratadine.
History
Rupatadine discovery, pre-clinical and clinical development was performed by J. Uriach y Cia, S. A., a Spanish pharmaceutical company. It was launched in 2003 in Spain by J. Uriach y Cia, S. A., with the brand name of Rupafin. The registration of the product is approved in 23 countries from the EU, 8 Central American countries, Brazil, Argentina, Chile, Turkey and 14 African countries.
Efficacy in humans
The efficacy of rupatadine as treatment for allergic rhinitis (AR) and chronic idiopathic urticaria (CIU) has been investigated in adults and adolescents (aged over 12 years) in several controlled studies, showing a rapid onset of action and a good safety profile even in prolonged treatment periods of a year.[3][4][5]
- Rupatadine (I) is an authorized antihistaminic agent and has IUPAC name 8-Chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4-piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine, CAS number 158876-82-5 for the free base and the following chemical formula:
![Figure imgb0001]()
- Rupatadine is currently marketed in 10 mg (rupatadine) tablets as rupatadine fumarate (CAS 182349-12-8 for the fumarate salt) for the treatment of allergic rhinitis and urticaria in adults and teenagers.
- Rupatadine free base was first disclosed in EP0577957 .
- Spanish patent application ES2087818 discloses the monofumarate salt of rupatadine (i.e. rupatadine fumarate) and aqueous liquid pharmaceutical compositions of rupatadine fumarate. In particular, this document discloses a syrup containing rupatadine fumarate at 4 g/L, sucrose, a flavouring agent, a sweetening agent and water; and a solution for injection which contains rupatadine fumarate at 20 g/L, benzyl alcohol, propyleneglycol and water.
- Despite the aqueous liquid pharmaceutical compositions disclosed in EP0577957and ES2087818 , the inventors have found that the solubility in water of rupatadine fumarate is 2.9 g/L (see Reference example 1) and therefore these prior art formulations may have stability problems due to supersaturation of rupatadine free base or rupatadine fumarate and would not be suitable for use as a medicament.
- CN101669901 and CN101669926 disclose liquid formulations of rupatadine free base using cyclodextrins to dissolve rupatadine.
- CN101669901 is directed to liquid formulations of rupatadine free base for ophthalmic delivery comprising rupatadine, a solvent and a cyclodextrin.
- CN10169926 is directed to liquid formulations of rupatadine free base for nasal delivery comprising rupatadine, a solvent and a cyclodextrin. It is stated that rupatadine has low solubility in water (1.39 mg/mL to 0.82 mg/mL at pH 3.0 to 7.0, table 9 in CN10169926 ) and the problem of its low solubility is solved using cyclodextrins (tables 10-12 of CN10169926 ) in order to obtain liquid formulations.



The reaction of 2-cyano-3-methylpyridine (I) with H2SO4 in t-BuOH gives the N-tert-butylamide (II), which is treated with two equivalents of BuLi and the corresponding dianion alkylated with 3-chlorobenzyl chloride to afford amide (III). The treatment of (III) with POCl3 gives nitrile (IV) which is cyclized to ketone (V) by subsequent treatment with CF3SO3H and aqueous HCl. Reaction of ketone (V) with the Grignard derivative prepared from chloride (VI) affords alcohol (VII) which is finally dehydrated by H2SO4 to give UR-12592 (1), as shown in Scheme 20491401a. The key intermediate (VI) is synthesized through the condensation of 5-methylnicotinic acid (VIII) with 4-hydroxypiperidine by means of DCC in DMF to give amide (IX), followed by reduction with POCl3 and NaBH4 to give the amino alcohol (X) which is treated with SOCl2. Scheme 20491402a. Description White crystals, m.p. 196-8 癈. References 1. Carceller, E., Jim閚ez, P.J., Salas, J. (J. Uriach & Cia SA). Process for the preparation of 8-chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4 -piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine. ES 9602107.

The key intermediate (VI) is synthesized through the condensation of 5-methylnicotinic acid (VIII) with 4-hydroxypiperidine by means of DCC in DMF to give amide (IX), followed by reduction with POCl3 and NaBH4 to give the amino alcohol (X), which is treated with SOCl2.![]()
………………………….
EXAMPLE 4
8-chloro-11-(1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
- To a solution of 1.7 mL (15 mmol) of 3,5-lutidine in 100 mL of CCl₄ was added 2.6 g (15 mmol) of NBS and the mixture was stirred at reflux under an argon atmosphere for 2 h. Then, the mixture was allowed to cool, the solid obtained was filtered off and to the filtrate was added 2.4 g (7.5 mmol) of the compound obtained in preparation 1 and 20 mg of 4-(dimethylamino)pyridine. The resulting mixture was stirred at room temperature for 18 h and 1.68 mL of triethylamine was added. It was diluted with 100 mL of dichloromethane and washed with 0.5N NaHCO₃ solution and with water. The organic phase was dried over sodium sulfate and the solvent was removed, to give 5.7 g of a residue that was chromatographed on silica gel (chloroform : methanol : ammonia, 60 : 2 : 0.2). 1.3 g of the title compound of the example was obtained as a white solid (yield: 40%).
mp: 58-61°C;
IR (KBr) ν: 3419, 3014, 1635, 1576, 1472 cm⁻¹;
¹H RMN (80 MHz, CDCl₃) δ (TMS): 8.39 (m, 3H, ar), 7.48 (m, 1H, ar), 7.37 (m, 1H, ar), 7.12 (m, 4H, ar), 3.45 (s, 2H, CH₂N), 3.36 (m, 2H), 3.1-2.1 (m, 13H). ¹³C RMN (20.15 MHz, CDCl₃) δ (TMS): 157.20 (C), 148.93 (CH), 147.46 (CH), 146.48 (CH), 139.50 (C), 138.56 (C), 137.06 (CH), 133.3 (C), 132.54 (C), 130.67 (CH), 128.80 (CH), 125.85 (CH), 121.92 (CH), 59.84 (CH₂), 54.63 (CH₂), 31.70 (CH₂), 31.32 (CH₂), 30.80 (CH₂), 30.56 (CH₂), 18.14 (CH₃).
………………………….
WO2006114676
Scheme-1

Example 1
Preparation of3-bromomethyl-5-methylpyridine hydrochloride: A mixture of carbon tetrachloride (4000ml), azobisisobutyronitrile (45.96gm, 0.279mol), 3,5-lutidine (150gm, 1.399mol) and N-bromosuccinamide (299.4gm, 1.682mol) is refluxed for 2 hours. The reaction mixture is cooled to room temperature and solid is filtered. HCl gas is passed to the filtrate and solid obtained is separated and filtered. Yield is 196gm Yield is 67.66%. Example 2
Preparation of Rupatadine :
A mixture of desloratadine (5.0gm, 0.016mol), methylene chloride (15ml), tetrabutylammonium bromide (0.575gm, 0.0018mol) and sodium hydroxide solution (2.5gm, 0.064mol in 8ml water) is cooled to 0 to 50C. 3-bromomethyl-5- methylpyridine hydrochloride (7.18gm, 0.032mol) in methylene chloride (35ml) is added to above mixture. The reaction mixture is stirred at 0 to 50C for 1 hour and at room temperature for 12 hours. Layers are separated and organic layer is washed with dilute HCl solution and water. Methylene chloride is distilled. Yield = 9.5g %Yield =
67.66%.
Example 3
Preparation of “Rupatadine fumarate:
A solution of fumaric acid (3.3gm) in methanol (46ml) is added to solution of
Rupatadine (4.5gm) in ethyl acetate (30ml) at room temperature. The reaction mass is cooled to -5 to O0C for 4 hours. Rupatadine fumarate is separated filtered and Washed with ethylacetate. Yield = 5.5 gm.
…………………………..
NEW PATENT
EP-02824103…An improved process for the preparation of rupatadine fumarate, Cadila Pharmaceuticals Ltd
Process for the preparing rupatadine intermediate (particularly 5-methylpyridine-3-methanol) comprises reduction of 5-methyl nicotinic acid alkyl ester using alkali metal borohydride is claimed. For a prior filing see WO2006114676, claiming the process for preparation of rupatadine fumarate.
……………………………………
J. Med. Chem., 1994, 37 (17), pp 2697–2703
DOI: 10.1021/jm00043a009
References
- Patents: EP 577957, US 5407941, US 5476856
- Merlos, M.; Giral, M.; Balsa, D.; Ferrando, R.; Queralt, M.; Puigdemont, A.; García-Rafanell, J.; Forn, J. (1997). “Rupatadine, a new potent, orally active dual antagonist of histamine and platelet-activating factor (PAF)”. The Journal of Pharmacology and Experimental Therapeutics 280 (1): 114–121. PMID 8996188.
- Picado, C. S. (2006). “Rupatadine: Pharmacological profile and its use in the treatment of allergic disorders”. Expert Opinion on Pharmacotherapy 7 (14): 1989–2001. doi:10.1517/14656566.7.14.1989. PMID 17020424.
- Keam, S. J.; Plosker, G. L. (2007). “Rupatadine: A review of its use in the management of allergic disorders”. Drugs 67 (3): 457–474. doi:10.2165/00003495-200767030-00008. PMID 17335300.
- Mullol, J.; Bousquet, J.; Bachert, C.; Canonica, W. G.; Gimenez-Arnau, A.; Kowalski, M. L.; Martí-Guadaño, E.; Maurer, M.; Picado, C.; Scadding, G.; Van Cauwenberge, P. (2008). “Rupatadine in allergic rhinitis and chronic urticaria”. Allergy 63: 5–28.doi:10.1111/j.1398-9995.2008.01640.x. PMID 18339040.
Literature References: Dual antagonist of histamine H1 and platelet-activating factor receptors. Prepn: E. Carceller et al., ES 2042421; eidem, US 5407941 (1993, 1995 both to Uriach);
eidem,J. Med. Chem. 37, 2697 (1994).
Mechanism of action: M. Merlos et al., J. Pharmacol. Exp. Ther. 280, 114 (1997).
Clinical trial in seasonal allergic rhinitis: F. Saint-Martin et al., J. Invest. Allergol. Clin. Immunol. 14, 34 (2004);
and comparison with ebastine: E. M. Guadaño et al., Allergy 59, 766 (2004).
Review of pharmacology and clinical development: N. Y. Van Den Anker-Rakhmanina, Curr. Opin. Anti-Inflam. Immunomod. Invest. Drugs 2, 127-132 (2000).
1 TO 8 OF 8 | ||
|---|---|---|
| PATENT | SUBMITTED | GRANTED |
| 8-chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine [US5407941] | 1995-04-18 | |
| Treatment of PAF and histamine-mediated diseases with 8-chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine [US5476856] | 1995-12-19 | |
| Process for the synthesis of n-(5-methylnicotinoyl)-4 hydroxypiperidine, a key intermediate of rupatadine [US6803468] | 2004-03-04 | 2004-10-12 |
| $g(b)2-ADRENERGIC RECEPTOR AGONISTS [EP1003540] | 2000-05-31 | |
| $g(b)2-ADRENERGIC RECEPTOR AGONISTS $g(b)2-ADRENERGIC RECEPTOR AGONISTS [EP1019360] | 2000-07-19 | |
| 8-Chloro-11-[1-[(5-methyl-3-pyridyl)methyl]-4-piperidyliden]-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridine. [EP0577957] | 1994-01-12 | 1995-07-12 |
| NOVEL CRYSTALLINE FORM OF RUPATADINE FREE BASE [US2009197907] | 2009-08-06 | |
| METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS METHODS FOR IDENTIFYING NOVEL MULTIMERIC AGENTS THAT MODULATE RECEPTORS [WO9966944] | 1999-12-29 | |
| |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 8-Chloro-6,11-dihydro-11-[1-[(5-methyl-3-pyridinyl)methyl]-4-piperidinylidene]-5H-benzo[5,6]cyclohepta[1,2-b]pyridine fumarate | |
| CLINICAL DATA | |
| TRADE NAMES | Rupafin, Alergoliber, Rinialer, Pafinur, Rupax, Ralif |
| AHFS/DRUGS.COM | International Drug Names |
| LEGAL STATUS |
|
| ROUTES | Oral |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | 98–99% |
| METABOLISM | Hepatic, CYP-mediated |
| HALF-LIFE | 5.9 hours |
| EXCRETION | 34.6% urine, 60.9% faeces |
| IDENTIFIERS | |
| CAS NUMBER | 158876-82-5 (free base) 182349-12-8 (fumarate) |
| ATC CODE | R06AX28 |
| PUBCHEM | CID 133017 |
| CHEMSPIDER | 117388 |
| UNII | 2AE8M83G3E |
| CHEMBL | CHEMBL91397 |
| CHEMICAL DATA | |
| FORMULA | C26H26ClN3 |
| MOLECULAR MASS | 415.958 g/mol |
↧
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide

N-[2-(7-Benzyl-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl)ethyl]acetamide
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide
Acetamide, N-[2-[7-(cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl]-
339.47, C22 H29 N O2
cas 1287785-08-3
Melatonin MT2 Agonists
Takeda……..innovator
Treatment of Sleep Disorders,
- Melatonin (N-acetyl-5-methoxytryptamine), which is a hormone synthesized and secreted principally in the pineal gland, increases in dark circumstances and decreases in light circumstances. Melatonin exerts suppressively on pigment cells and the female gonads, and acts as a synchronous factor of biological clock while taking part in transmittance of photoperiodic code. Therefore, melatonin is expected to be used for the therapy of diseases related with melatonin activity, such as reproduction and endocrinic disorders, sleep-awake rhythm disorders, jet-lag syndrome and various disorders related to aging, etc.
- Recently, it has been reported that the production of melatonin melatonin could reset the body’s aging clock (see Ann. N. Y. Acad. Sci., Vol. 719, pp. 456-460 (1994)). As previously reported, however, melatonin is easily metabolized by metabolic enzymes in vivo (see Clinical Examinations, Vol. 38, No. 11, pp. 282-284 (1994)). Therefore, it cannot be said that melatonin is suitable as a pharmaceutical substance.
- Various melatonin agonists and antagonists such as those mentioned below are known.
- (1) EP-A-578620 discloses compounds of:
![Figure 00020001]()
- (2) EP-A-420064 discloses a compound of:
![Figure 00020002]()
- (3) EP-A-447285 discloses a compound of:
![Figure 00020003]()
- (4) EP-A-662471 discloses a compound of:
![Figure 00020004]()
- (5) EP-A-591057 discloses a compound of:
![Figure 00020005]()
- (6) EP-A-527687 discloses compounds of:
![Figure 00030001]() X=S, 0, Y=CH
X=S, 0, Y=CH
X=0, NH, Y=N - (7) EP-A-506539 discloses compounds of:
![Figure 00030002]()
- Tricyclic or more poly-cyclic compounds with a cyclic ether moiety, such as those mentioned below, are known.
- (1) Compounds of:
![Figure 00030003]() are disclosed in Tetrahedron Lett., Vol. 36, p. 7019 (1995).
are disclosed in Tetrahedron Lett., Vol. 36, p. 7019 (1995). - (2) Compounds of:
![Figure 00040001]()
![Figure 00040002]() are disclosed in J. Med. Chem., Vol. 35, p. 3625 (1992).
are disclosed in J. Med. Chem., Vol. 35, p. 3625 (1992). - (3) Compounds of:
![Figure 00040003]() are disclosed in Tetrahedron, Vol. 48, p. 1039 (1992).
are disclosed in Tetrahedron, Vol. 48, p. 1039 (1992). - (4) Compounds of:
![Figure 00040004]() are disclosed in Tetrahedron Lett., Vol. 32, p. 3345 (1991).
are disclosed in Tetrahedron Lett., Vol. 32, p. 3345 (1991). - (5) A compound of:
![Figure 00050001]() is disclosed in Bioorg. Chem., Vol. 18, p. 291 (1990).
is disclosed in Bioorg. Chem., Vol. 18, p. 291 (1990). - (6) A compound of:
![Figure 00050002]() is disclosed in J. Electroanal. Chem. Interfacial Electrochem., Vol. 278, p. 249 (1990).
is disclosed in J. Electroanal. Chem. Interfacial Electrochem., Vol. 278, p. 249 (1990).
see - (1) Compounds of:














Highly Potent MT2-Selective Agonists
J. Med. Chem., 2011, 54 (9), pp 3436–3444
DOI: 10.1021/jm200221q
N-{2-[7-(Cyclohexylmethyl)-1,6-dihydro-2H-indeno[5,4-b]furan-8-yl]ethyl}acetamide (15)
By a similar procedure that described for 8, 15 (79%) was obtained as a white solid; mp 133−134 °C (EtOAc/hexane).
1H NMR (CDCl3) δ 0.82−1.03 (2H, m), 1.06−1.32 (3H, m), 1.42−1.78 (6H, m), 1.96 (3H, s), 2.32 (2H, d, J = 7.2 Hz), 2.74 (2H, t, J = 7.2 Hz), 3.26 (2H, s), 3.32−3.52 (4H, m), 4.59 (2H, t, J= 8.6 Hz), 5.60 (1H, s), 6.59 (1H, d, J = 7.9 Hz), 7.11 (1H, d, J= 7.9 Hz).
MS (ESI) m/z 340 (M + H)+. Anal. (C22H29NO2) C, H, N.
↧
LY-156735 (TIK-301, PD-6735)….for the treatment of sleep latency in patients with primary insomnia
LY-156735 (TIK-301, PD-6735)….for the treatment of sleep latency in patients with primary insomnia
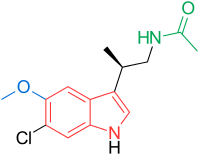
N-[(2R)-2-(6-chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide
LY-156735 (TIK-301, PD-6735) is a melatonin MT1 and MT2 agonist which is under development for the treatment of insomnia and other sleep disorders.[1]
Beta-methyl-6-chloromelatonin (PD-6735) is a melatonin MT1 and MT2 agonist which had been in phase II trials at Phase 2 Discovery for the treatment of sleep latency in patients with primary insomnia, however, no recent development has been reported.
The melatonin agonist exhibits high selectivity and provides a novel mode of action different from that of benzodiazepine receptor ligands currently on the market.
Furthermore, the drug candidate is believed to be non-addicting, therefore, offering an advantage over marketed sleep medications. Originally discovered by Lilly, PD-6735 was licensed to Phase 2 Discovery in 2002 for further development.
Orphan drug designation has been assigned in the U.S. for the treatment of circadian rhythm sleep disorders in blind people with no light perception and for the treatment of neuroleptic-induced tardive dyskinesia in schizophrenia patients.
In 2007, the product candidate was licensed to Tikvah Therapeutics by Phase 2 Discovery for worldwide development and commercialization for the treatment of sleep disorder, depression and circadian rhythm disorder.
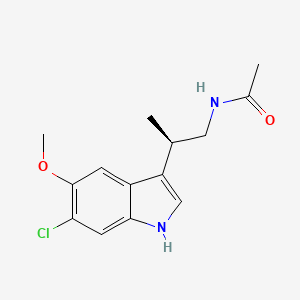
beta -alkylmelatonins as ovulation inhibitors [US4997845]1991-03-05
BETA-ALKYLMELATONINS [EP0281242]1988-09-07 GRANT1992-08-12

The condensation of 6-chloro-5-methoxy-1H-indole (I) with Meldrum’s acid (II) and acetaldehyde (III) catalyzed by L-proline in acetonitrile gives the adduct (IV), which is treated with Cu and ethanol in refluxing pyridine to yield 3-(6-chloro-5-methoxy-1H-indol-3-yl)butyric acid ethyl ester (V). The reaction of (V) with hydrazine at 140 C affords the hydrazide (VI), which is treated with NaNO2 and Ac-OH to provide the corresponding azide that, without isolation, is thermolyzed and rearranged in toluene at 80?C to give 7-chloro-6-methoxy-4-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (VII). The cleavage of the lactam ring of (VII) with KOH in refluxing ethanol/water yields 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (VIII). The decarboxylation of (VIII) by means of refluxing aq. 3M HCl affords 3-(2-amino-1-methylethyl)-6-chloro-5-methoxy-1H-indole (IX), which is finally acylated with acetic anhydride and pyridine in toluene to provide the target 6-chloromelatonin as a racemic compound.![]()
EP 0281242;……….http://www.google.com/patents/EP0281242B1?cl=en
Example 3
- Preparation of β-Methyl-6-chloromelatonin
- Following the procedure of Example 1, a solution of 10.0 g (0.055 mole) of 5-methoxy-6-chloroindole, 3.1 ml (2.44 g, 0.055 mole) of acetaldehyde, and 7.94 g (0.055 mole) of Meldrum’s acid in 90 ml of acetonitrile was stirred for 48 hours. The solvent was removed under vacuum, and the adduct thus prepared was recrystallized by dissolving in warm toluene and immediately cooling. The adduct was obtained as slightly pink crystals; m.p. = 145°C; yield = 16.5 g (85%). The elemental analysis of the product showed a slightly elevated percentage of carbon. However, the NMR spectrum indicated that the product was pure and had the indicated structure.
Analysis calc. for C₁₇H₁₈NO₅Cl- Theory:
- C, 58.04; H, 5.16; N, 3.98; Cl, 10.08
- Found :
- C, 59.34; H, 5.15; N, 3.84; Cl, 9.69
- The solvolysis and decarboxylation of the adduct (11.0 g; 31.3 mmoles) using ethanol, pyridine, and copper dust was carried out by the procedure of Example 1. The yield of 3-(5-methoxy-6-chloro-1H-indol-3-yl)pentanoic acid ethyl ester, a pale yellow oil, after chromatography over silica gel using 10% EtOAc/90% toluene was 8.68 g (94%).
Analysis calc. for C₁₅H₁₈NO₃Cl- Theory:
- C, 60.91; H, 6.13; N, 4.74; Cl, 11.99
- Found :
- C, 60.67; H, 5.86; N, 4.93; Cl, 11.73
- A mixture of 8.68 g (29.3 mmoles) of the above ethyl ester and 6 ml of hydrazine hydrate was heated at 140°C under nitrogen in a flask fitted with an air cooled condensor. After 6½ hours, the excess hydrazine hydrate was removed under vacuum. The 2-methyl-2-(5-methoxy-6-chloro-3-indolyl)-propionhydrazide thus prepared was recrystallized from ethyl acetate; Yield = 7.13 g (86%); m.p. = 154-155°C.
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.14; H, 5.51; N, 14.49; Cl, 12.78
- The above hydrazide (7.13 g, 25 mmoles) was converted to the corresponding acyl azide, the azide thermolyzed and rearranged at 80° in toluene, and the rearranged product cyclized with HCl according to the procedure of Example 1. The yield of crude, light tan, lactam, 1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole, product, (m.p. = 249-252°C) was 4.77 g (72%).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58
- Found :
- C, 59.45; H, 4.77; N, 10.72
- The crude lactam (4.77 g, 18 mmoles) was hydrolyzed with aqueous ethanolic KOH as described in Example 1. The yield of crude amino acid, 2-carboxy-3-(1-amino-2-propyl)-5-methoxy-6-chloroindole, was 3.98 g (78%). The crude product (3.0 g; 10.6 mmoles) was decarboxylated, using the procedure of Example 1, by refluxing in 100 ml of 3M HCl overnight. The acidic solution was decolorized with activated carbon and was then basified with 5M NaOH. The amine was extracted into diethyl ether. After drying the ether extract over Na₂SO₄, the diethyl ether was removed in vacuoleaving as a residue the crystallized tryptamine, 3-(1-amino-2-propyl)-5-methoxy-6-chloroindole; m.p. 133-4°C. The yield, after recrystallization from toluene/hexane, was 1.62 g (64%).
Analysis calc. for C₁₂H₁₅N₂OCl- Theory:
- C, 60.38; H, 6.33; N, 11.74; Cl, 14.85
- Found :
- C, 60.11; H, 6.05; N, 11.93; Cl, 15.06
- A solution of 1.51 g (6.3 mmoles) of the above tryptamine in 10 ml of toluene and 2.5 ml of pyridine was treated with 1.5 ml of acetic anhydride. After allowing the reaction mixture to stand for three hours at room temperature, the volatile materials were removed under vacuum. The residue was dissolved in ethyl acetate, and washed with aqueous NaHCO₃, and brine. The ethyl acetate solution was dried over Na₂SO₄, and the solvent removed by evaporation. The residual oil was crystallized from toluene/hexane yielding 6-chloro-β-methylmelatonin, (m.p. = 133-5°C; 1.09 g, 61%).
Analysis calc. for C₁₄H₁₇N₂O₂Cl- Theory:
- C, 59.89; H, 6.10; N, 9.98; Cl, 12.63
- Found :
- C, 60.03; H, 6.22; N, 9.75; Cl, 12.92
…………………………………………….
PATENT

The intermediate diazonium salt (XIII) has been obtained as follows: the hydrogenation of 3-chloro-4-methoxynitrobenzene (XI) with H2 over Pt/Al2O3 in toluene gives the corresponding aniline (XII), which is diazotized with NaNO2/HCl and treated with sodium tetrafluoroborate to yield the target diazonium salt intermediate (XIII). The reduction of pulegone (I) with H2 over Pd/C gives the menthol (II), which is oxidized with CrO3/H2SO4 to yield 3(R),7-dimethyl-6-oxooctanoic acid (IV), which can also be obtained by direct oxidation of (l)-menthol (III) under the same conditions.
The oxidation of (IV) with trifluoroperacetic acid (trifluoroacetic anhydride/H2O2) in dichloromethane yields the 3(R)-methylhexanedioic acid isopropyl monoester (V), which is treated with NaOEt in ethanol to obtain the corresponding ethyl monoester (VI). The reaction of (VI) with diethyl carbonate, EtONa, and “Adogen 464″ (a phase transfer catalyst) in ethanol affords 5,5-bis(ethoxycarbonyl)-3(S)-methylpentanoic acid (VII), which is treated with oxalyl chloride to provide the expected acyl chloride (VIII). The reaction of (VIII) with sodium azide and benzyl alcohol gives the intermediate azide that rearranges to the benzyl carbamate (IX).
The reductive cyclization of (IX) with H2 over Pd/C in ethanol yields 5(R)-methyl-2-oxopiperidine-3-carboxylic acid ethyl ester (X), which is condensed with the intermediate diazonium salt (XIII) to afford the hydrazono derivative (XIV). The cyclization of (XIV) in hot formic acid provides 7-chloro-6-methoxy-4(R)-methyl-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indol-1-one (XV), which is treated with KOH In refluxing ethanol/water to cleave the lactam ring, yielding 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole-2-carboxylic acid (XVI). The decarboxylation of (XVI) by means of refluxing 3M HCl affords 3-(2-amino-1(R)-methylethyl)-6-chloro-5-methoxy-1H-indole (XVII), which is finally acylated with Ac2O and pyridine in toluene to provide the target 6-chloromelatonin as a pure enantiomer.![]()
Example 7
- Preparation of S-(-)-β-methyl-6-chloromelatonin and R-(+)-β-methyl-6-chloromelatonin
- A solution of 4.0 g (21 mmoles) of 3-chloro-4-methoxynitrobenzene in 200 ml of toluene was hydrogenated over 0.4 g of 5% platinum on alumina. The catalyst was removed by filtration and the solvent evaporated from the filtrate. The crude 3-chloroanisidine prepared was placed in solution in diethyl ether and treated with ethereal HCl to produce the hydrochloride salt, which was collected and dried; weight = 2.48 g (61% yield).
- A mixture of 2.40 g (12.4 mmoles) of 3-chloroanisidine hydrochloride in 7 ml of 4M HCl was treated, at 0°C, with 0.86 g (12.5 mmoles) of sodium nitrite in 5 ml of water. After stirring at 0°C for an hour the solution was filtered and the filtrate added slowly to an ice cold solution of 2.6 g (24 mmoles) of sodium fluoroborate in 8 ml of water. After stirring at 0°C for an hour the salt was collected and washed successively with, cold 5% sodium fluoroborate solution, cold methanol, and ether. The dried 3-chloro-4-methoxybenzene diazonium fluoroborate thus prepared weighed 2.2 g (69% yield).
- A mixture of 2.03 g (11.0 mmole) of (R)-(-)-3-ethoxycarbonyl-5-methyl-2-piperidone and 30 ml of 0.75M NaOH was stirred at room temperature (24°C) overnight. The solution was cooled to 0°C and the pH lowered to 3.5 with 3M hydrochloric acid. The diazonium salt (2.8 g, 10.9 mmoles) was added in small portions and the reaction mixture cooled to about 0°C overnight. The product, R-(-)-3-(3-chloro-4-methoxy)phenylhydrazono-5-methyl-2-piperidone, was collected, washed with water, and dried; weight = 2.30 g (75% yield); m.p. = 205°C. A small sample was further purified by chromatography over a short silica gel column using ethyl acetate as the eluant. [α]²⁵ = -58° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₆N₃O₂Cl- Theory:
- C, 55.42; H, 5.72; N, 14.91; Cl, 12.58
- Found :
- C, 55.79; H, 5.78: N, 14.72; Cl, 12.69
- A mixture of 2.20 g (7.8 moles) of the R-(-) hydrazone and 20 ml of 90% formic acid was heated at 85° for three hours then slowly diluted with an equal volume of water. The mixture was allowed to cool and then chilled overnight. The dark precipitate was collected, washed with water, then recrystallized from acetone/water, yielding 1.20 g (60% yield) of S-(-)-1-oxo-4-methyl-6-methoxy-7-chloro-1,2,3,4-tetrahydro-9H-pyrido[3,4-b]indole; m.p. = 248°C. [α]²⁵ = -12.2° (c = 10, MeOH).
Analysis calc. for C₁₃H₁₃N₂O₂Cl- Theory:
- C, 58.99; H, 4.95; N, 10.58; Cl, 13.39
- Found :
- C, 59.16; H, 4.88; N, 10.80; Cl, 13.15
- The conversion of (S)-(-)-lactam to (S)-(-)-6-chloro-β-methylmelatonin was carried out as described previously in Example 3. The product, S-(-)-β-methyl-6-chloromelatonin, was spectroscopically identical to the racemate, but gave an optical rotation of [α]²⁵ = -13.2° (c = 10, MeOH).
- (R)-(+)-6-chloro-β-methylmelatonin was synthesized from (S)-(+)-3-ethoxycarbonyl-5-methyl-2-piperidone in the same manner as described above. The stereoisomer was identical to the (S)-(-) material except for the sign of rotation.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| N-[(2R)-(6-Chloro-5-methoxy-1H-indol-3-yl)propyl]acetamide | |
| CLINICAL DATA | |
| LEGAL STATUS | ? |
| IDENTIFIERS | |
| CAS NUMBER | 118702-11-7 |
| ATC CODE | ? |
| PUBCHEM | CID 219018 |
| CHEMSPIDER | 189853 |
| CHEMICAL DATA | |
| FORMULA | C14H17ClN2O2 |
| MOLECULAR MASS | 280.757 |
References
↧
↧
Antimalarial Drug TCMDC 123812 and 123794
Synthesis of Analogs of Arylpyrrole Antimalarial Drug Leads
Abstract
Malaria continues to be one of the most widespread infectious diseases and with recent focus on global eradication and the continual evolution of drug resistant parasitic strains, the search for potent new antimalarials has gained momentum. In contrast to rational drug design approaches, the high-throughput screening of large compound libraries for potency against the parasite in whole cell assays has identified the Arylpyrrole series as a promising chemical lead. Before looking to comprehend the complex modes of biological action, the evaluation of analogs within this new drug class and the validation of their organic synthesis may act to identify specific compounds with increased potency or chemical moieties that lend themselves to the optimisation of the drug synthesis process. The collaborative efforts of open source drug discovery may help to identify potent compounds with a high-yielding and cost effective manufacturing process and thus accelerate the development of a promising candidate for integration into clinical practice.
Introduction
Malaria is a widespread tropical infectious disease caused by a number of species of the protozoan parasite of the genus plasmodium, most notably P. falciparum, which are transmitted through the saliva of the anopheles mosquito. Per year, more than 240 million people suffer from the disease and approximately 40% of the global population live in countries where Malaria is endemic.[1] As both a consequence of and a great contributor towards poverty, Malaria costs sub-Saharan Africa alone US$12 billion in productivity every year and while an approximate cost limitation for one antimalarial treatment is US $1[1], current estimates suggest that many may only be able to afford 10 cents per treatment.[2] Thus with the majority of sufferers of a poor socioeconomic background, drug design should focus on the cost-effective synthesis of highly potent compounds, in order to minimise the quantities administered.
Driven by a knowledge of the biology of the parasite, conventionally rational drug design deduces leads based on first identifying a possible molecular target and the potential mechanisms of drug action at that target. However through reversing this process, a high-throughput screening for potent compounds in whole cell assays can act to expedite the lead identification process.[3] By this approach a single compound may be applicable to many ambiguous or poorly understood, more complex biological targets, decreasing the chances of a parasitic strain evolving resistance mechanisms at every target. Ultimately this approach will allow the rapid identification of leads which, because of their capacity to act on complex targets, may result in more effective prophylactic or therapeutic drugs which retain their activity for longer.
Recently the data from the high-throughput screening of a two million compound library was released by GlaxoSmithKline (GSK), identifying 13, 533 compounds, which inhibited the intra-erythrocytic cycle of multidrug resistant strains of P. falciparum by more than 70%. This identified several promising classes of compounds which retained low cytotoxicity to human cell lines.[4] [5] Among these, the arylpyrrole series has been isolated and the resynthesis and validation of a number of lead compounds within this series are being conducted using open source science. Disseminating information over many online mediums has encouraged collaborative efforts within the international scientific community and this free exchange of data and ideas between organisations, individuals and academic institutions may stimulate faster optimisation and development of leads.
Initial validation of arylpyrrole leads involved the resynthesis of two known series compounds, that is TCMDC 123812 and TCMDC 123794, as high quality starting points for lead optimisation with reasonable druggability.[6] Following evaluation of two possible synthesis pathways to these final compounds, slight changes were introduced to the chemical substructures and synthesis re-evaluated. Based on knowledge of the in vivo functionality of certain moieties, analogs were identified that may influence synthesis or beneficially contribute to the overall drug profile and efficacy.
Modifications to the para-position of aryl ring were evaluated, with hydrogen, methyl and trifluoromethyl groups being chosen to substitute the fluorine present in the original compounds. The introduction of bonds weaker than the original carbon-fluorine bond at that position may influence metabolic stability, influencing the affinity of the drug to para-hydroxylation.[7] However, and especially in the instance of a hydrogen in that position, a null result may be invaluable in enabling the prioritisation of other more stable functional groups. Thus antiplasmodial activity and cytotoxicity may be impacted upon as variations may influence the modes of action, changing the in vivo efficacy of the drug. Resultant changes in the physical or chemical properties of analogs may affect the potential for developing high-yielding, efficient and cost effective synthesis strategies. However ramifications of these chemical modifications on the ease of organic synthesis are hard to predict and thus the viability of the synthesis of a range of analogs must be evaluated as primary steps in the lead optimisation process.
Two potential synthesis pathways were identified for obtaining the carboxylic acid which then undergoes the addition of an amide group through a coupling reaction to achieve the final compound.[6] The first relied on a Paal Knorr cyclisation reaction between a dicarbonyl compound and a para-substituted aniline resulting in the arylpyrrole core. A Vilsmeier-Haack reaction then employed the use of phosphoryl chloride and dimethylformamide to form a Vilsmeier reagent or chloroimminium ion, which when substituted onto the pyrrole ring acts as a formylating agent to produce an aldehyde, once hydrolysed at a low pH. A second synthesis pathway involved synthesis of an ester, initially involving alkylation using a reactive enolate ion to form an intermediate before continuing with a condensation reaction into the characteristic arylpyrrole core. This report will describe the synthesis of each analog via these two alternative synthesis pathways. (Fig. 1)
Materials and Methods
1. Paal Knorr Synthesis of 2,5-dimethyl-1H-phenyl-pyrrole
2. Paal Knorr synthesis of 2,5-dimethyl-1H-(p-tolyl)- pyrrole
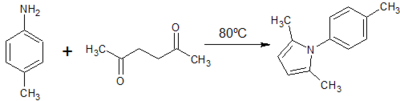
3. Paal Knorr Synthesis of 2,5-dimethyl -1H-(p-trifluoromethyl)phenyl-pyrrole
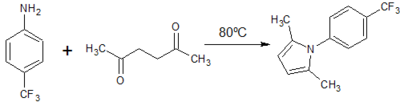
4. Vilsmeier-Haack synthesis of 2,5-dimethyl-1H-phenyl pyrrole-3-carboxaldehyde

5. Vilsmeier-Haack Synthesis of 2,5-dimethyl-1H-(p-tolyl)-pyrrole-3-carboxaldehyde.
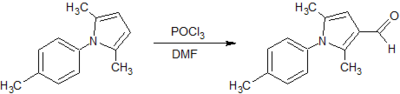
6. Synthesis of ethyl 2,5-dimethyl-1-(p-tolyl)-1H-pyrrole-3-carboxylate
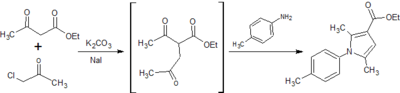
7. Synthesis of ethyl 2,5-dimethyl-1-phenyl-1H-pyrrole-3-carboxylate
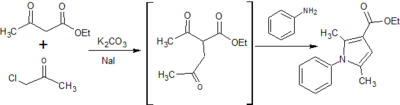
8. Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate
![Figure 9: Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate](http://openwetware.org/images/thumb/f/fe/LMW_9-1.png/400px-LMW_9-1.png)
General Reaction Mechanisms
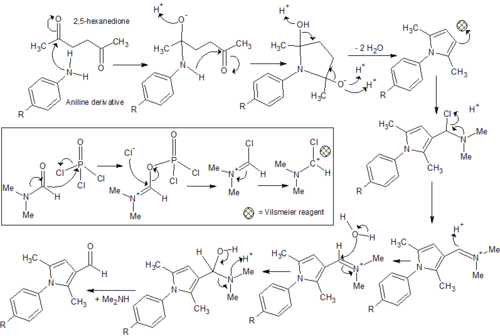

Results
1. Paal-Knorr Synthesis of 2,5-dimethyl-1H-phenyl-pyrrole
Yield: 6.2 g (60.4%)
Comment: Product crystallised easily. Reasonably pure red brown granular substance after recrystallization.
2. Paal-Knorr synthesis of 2,5-dimethyl-1H-(p-tolyl)- pyrrole
Yield: 8.3 g (74.5%)
Comment: Crystallisation required use of activated charcoal. Recrystallisation produced ochre-coloured sand-like, relatively pure compound at a reasonable yield.
3. Paal-Knorr Synthesis of 2,5-dimethyl -1H-(p-trifluoromethyl)phenyl-pyrrole
Yield: 6.08g (60%)
Comment: Reaction took slightly longer. Recrystallisation produced a relatively pure red-brown sand-like compound.
4. Vilsmeier-Haack synthesis of 2,5-dimethyl-1H-phenyl pyrrole-3-carboxaldehyde
Yield: 407 mg (64%)
Comment: Vilsmeier reaction completed reasonably quickly. Pure compound that crystallised easily produced after recrystallization. (grey/beige free-flowing powder)
5. Vilsmeier-Haack Synthesis of 2,5-dimethyl-1H-(p-tolyl)-pyrrole-3-carboxaldehyde.
Yield: 423 mg (71%)
Comment: Recrystallised fairly easily producing a fairly pure grey/brown lumpy powder.
6. Synthesis of ethyl 2,5-dimethyl-1-(p-tolyl)-1H-pyrrole-3-carboxylate
Yield: 1.8 g (45.6%)
Comment: Upon the use of both activated charcoal and column chromatography, product crystallised easily to a relatively pure bright yellow powder.
7. Synthesis of ethyl 2,5-dimethyl-1-phenyl-1H-pyrrole-3-carboxylate
Yield: 952 mg (approximate yield 37%)
Comment: Lack of separation between product and impurity in column chromatography resulted in product not crystallising.
8. Synthesis of ethyl 2,5-dimethyl-1-[p-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxylate
Yield: 1.85 g (55%)
Comment: Crystallised easily following column chromatography to form a pale yellow powder.
Discussion
Synthesis via each reaction pathway was viable for most analogs however procedures need to be optimised and due to the nature of the corresponding reactions to the carboxylic acid, synthesis via the ester could be considered the most favourable.
The synthesis of the aldehyde via a Vilsmeier Haack synthesis following a Paal Knorr cyclisation was viable and the products synthesised achieved moderate yields and a reasonable level of purity with minimal purification steps. While viable, procedures could still be optimised further, however the oxidation of the aldehyde to obtain the carboxylic acid has proved problematic for the original resynthesis using the p-fluoro-phenyl-pyrrole-carbaldehyde. Unless a forgiving oxidation reaction becomes apparent, the simple base-hydrolysis via saponification necessary for the ester could prove a more favourable approach. For this reason only two aldehydes were synthesised with this strategy.
Although the ester exists in the correct oxidation state which acts to mitigate further issues with the synthesis of the carboxylic acid, complications in purification steps need to be overcome before the synthesis of analogs could be considered practical using this pathway. Potassium carbonate acts to remove a labile hydrogen off the alpha carbon of ethyl acetoacetate, which forms a reactive enolate ion. Sodium iodide substitutes the chlorine of the chloroacetone, which becomes susceptible to nucleophilic attack and an alkylation reaction follows to form a reactive intermediate. Enolate ion formation is susceptible to side reactions such as alkylation at the oxygen and while alkylation of the alpha carbon is preferential, the synthesis of the intermediate may need to be refined to ensure minimal side products and impurities. Additionally immediate condensation of the crude intermediate with the aniline derivatives may minimise formation of further side products. All three analogs required column chromatography, resulting in a significant amount of product loss and diminishing from the simplicity and practicality required for large-scale drug manufacture. It must be noted, however, this condensation reaction proved forgiving in the resynthesis of the original compounds, achieving a reasonable yield with minimal purification steps.[8] Thus appropriate optimisation of the synthesis procedures could affirm this as the most practical pathway for each analog.
Given synthesis complications and a likely propensity for para-hydroxylation, it may be unlikely that the N-phenyl analogs present promising drug leads, however further synthesis trials and testing against the parasite are required to conclusively determine this. Aqueous extraction of the final ester was problematic as a result of increased miscibility with dilute citric acid, and unsuccessful separation of product and impurity in column chromatography prevented crystallisation. It is unclear at this stage whether the parasubstituted methyl or trifluoromethyl groups retain any steric, inductive or other effects that contribute to any physicochemical properties that lend themselves to improving organic synthesis or in vivo drug targeting. The synthesis strategies require optimisation to eliminate time consuming and costly purification steps and the proceeding hydrolysis and coupling reactions need validation before testing the in vivo efficacy of the drugs in whole cell assays. Alternative reaction conditions, reagents, solvents or even new synthesis pathways should be tested to achieve optimisation as, irrespective of the drug’s antiplasmodial activity, it is crucial that cost-effective simple procedures be conserved so that large-scale production and distribution to impoverished nations remains possible.
Following the validation and testing of current analogs, additional analogs may be evaluated based on informed decisions of the potential advantages of specific chemical groups and the success of similar moieties. Once a promising lead has been identified and validated, the mode of action and how the drug acts to mediate host parasite-interactions on a molecular level may be researched and the drug tailored for that specific functionality. Mechanisms of action have already been postulated for related arylpyrroles, informing the choice of analogs and pursuit of specific lead compounds.[9]
Open source science has contributed significantly to the rapid identification of promising drug leads. Contributions from a variety of fields of expertise allow both synthetic and biological concerns for these lead compounds to be addressed, giving a more holistic view of the nature of drug design goals. Stimulating discussion and participation, this free exchange of ideas enables involvement of the wider scientific community, accelerating research and development so that it may be possible to find an effective solution to a devastating problem as soon as possible.
References
- Wells, T.N.C., Alonso, P.L.and Gutteridge, W.E. “New Medicines to improve control and contribute to the eradication of Malara”, Nature Reviews,2009, Vol. 8, pp879-891
- Panosian, CB. “Economic access to effective drugs for falciparum malaria”, Clinical Infectious Diseases, 2005, Vol. 40, no. 5, pp. 713-717
- Wells T.N.C “Is the tide turning for New Malaria Medicines”, Science, 2010, Vol. 329 pp.1153-1154
- Francisco-Javier, G., Sanz, LM., Jaume, V. “Thousands of chemical starting points for antimalarial lead identification”, Nature, 2010, Vol. 465, no. 7296, pp305-U56
- Calderon, F., Barros, D., Beuno Jose, M. et al. “An Invitation to Open Innovation in Malaria Drug Discovery: 47 Quality Starting Points from the TCAMS”, ACS Chemistry Letters, 2011, Vol.2, no.10, pp741-746
- The Synaptic Leap – Malaria Research Community (Updated 2011)
- Todd, M., Ylioja, P. (2011). Open Source Drug Discovery – Malaria. Open wet ware.
- Lab Blog: Malaria.ourexperiment.org blog (Updated 2011)
- Lee, BH., Lee, MJ., Park, S,. Oh, DC,. Elsasser, S,. Chen, PC,. Gartner, C,. Dimova, N,. Hanna, J,. Gygi, SP,. Wilson, SM,. King, RW,. Finley, D. “Enhancement of proteasome activity by a small-molecule inhibitor of USP14”, Nature, 2010, Vol. 467, pp179-184
↧
Route Design in the 21st Century: The ICSYNTH Software Tool as an Idea Generator for Synthesis Prediction

The new computer-aided synthesis design tool ICSYNTH has been evaluated by comparing its performance in predicting new ideas for route design to that of historical brainstorm results on a series of commercial pharmaceutical targets, as well as literature data. Examples of its output as an idea generator are described, and the conclusion is that it adds appreciable value to the performance of the professional drug research and development chemist team.

† Chemical Development, AstraZeneca R&D, Silk Road Business Park, Macclesfield, SK10 2NA Cheshire, U.K.
‡ Chemnotia AB, Forskargatan 20 J, 151 36 Södertälje,Sweden
§ InfoChem GmbH, Landsberger Straße 408/V, D-81241 München, Germany
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/op500373e
Publication Date (Web): January 22, 2015
Copyright © 2015 American Chemical Society
Currently, ICSYNTH has assumed a place as a unique predictive tool for route design in Chemical Development in AZ. While it is finding valuable commercial application in our own and others’ hands, it remains a work in progress.



 InfoChem will be represented at the forthcoming ACS Meeting in San Diego. You will find Dr. Josef Eiblmaier, Dr. Valentina Eigner Pitto, and Dr. Peter Loew …
InfoChem will be represented at the forthcoming ACS Meeting in San Diego. You will find Dr. Josef Eiblmaier, Dr. Valentina Eigner Pitto, and Dr. Peter Loew …
ICSYNTH
SEE…………….http://www.slideshare.net/Haxel/icic-2014-knowledgebased-de-novo-molecular-design-using-icsynth-frp
InfoChem’s ICSYNTH is a powerful computer aided synthesis design tool that enables chemists to generate synthetic pathways for a target molecule. The benefit is that ICSYNTH can facilitate innovation by stimulating ideas for alternative or novel synthetic routes that otherwise may not be considered. This may lead to improved route design, for example shorter pathways or more economical reaction modifications.
After inputting the target, users can select different synthetic strategies depending on requirements. ICSYNTH then automatically generates a multistep interactive synthesis tree – each node on the tree representing a precursor. The advantages are that the suggested reactions are based on, and linked to, published reactions (or their analogs) and the precursor availability is automatically checked in commercial catalogs. Users can modify the synthesis tree or select precursors for further analysis.
At the heart of ICSYNTH is an algorithmic chemical knowledge base of transform libraries that are automatically generated from reaction databases. The number of transform libraries is only limited by the availability of validated reaction databases.
In addition to retro synthesis design, ICSYNTH has a forward reaction prediction module that offers reactivity mapping for the target molecule.Version 2.0 of ICSYNTH was launched in April 2014. The completely re-designed user interface (based on JavaScript) and major improvements in the algorithm responsible of the precursor search are the main enhancements of Version 2.0. In addition the forward reaction prediction algorithm has been optimized. Click here to see a complete version history.
In addition to retro synthesis design, ICSYNTH has a forward reaction prediction module that offers reactivity mapping for the target molecule.Version 2.0 of ICSYNTH was launched in April 2014. The completely re-designed user interface (based on JavaScript) and major improvements in the algorithm responsible of the precursor search are the main enhancements of Version 2.0. In addition the forward reaction prediction algorithm has been optimized. Click here to see a complete version history.





INFOCHEM GESELLSCHAFT FÜR CHEMISCHE INFORMATION MBH
Landsberger Straße 408/V
D-81241 München
Germany
D-81241 München
Germany
| PHONE: | +49 (0)89 58 30 02 |
|---|---|
| FAX: | +49 (0)89 580 38 39 |
| EMAIL: | info@infochem.de |



 Historische Bilder der Landsberger Straße – An der Trambahnhaltestelle Holzapfelstraße endet – Münchner Straßen – München – Süddeutsche.de
Historische Bilder der Landsberger Straße – An der Trambahnhaltestelle Holzapfelstraße endet – Münchner Straßen – München – Süddeutsche.de↧
ANTHONY CRASTO VENTURES INTO CHINA.....MY KAIXIN BLOG 开心网 ON MEDICINAL CHEMISTRY
KAIXIN
![]()
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY...............http://www.kaixin001.com/home/?_profileuid=159073878
![]()
![]() CHINA
CHINA
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY...............http://www.kaixin001.com/home/?_profileuid=159073878

 CHINA
CHINAMY EASTERN VENTURE TO PROPAGATE CHEMISTRY...............http://www.kaixin001.com/home/?_profileuid=159073878
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY...............http://www.kaixin001.com/home/?_profileuid=159073878
MY EASTERN VENTURE TO PROPAGATE CHEMISTRY...............http://www.kaixin001.com/home/?_profileuid=159073878\


↧